











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
El síndrome de Andersen-Tawil (SAT) es una entidad genética rara que se caracteriza por la presencia de arritmias ventriculares, rasgos dismórficos a nivel esquelético y parálisis muscular periódica. El diagnóstico usualmente es tardío dada la presentación atemporal de las manifestaciones 1. El apoyo genético es crucial 2 y el tratamiento incluye manejo de la parálisis periódica, antiarrítmicos y uso de dispositivos para prevención de muerte súbita. A continuación se presenta un caso que se considera relevante dada prevalencia del síndrome, el retraso en el diagnóstico y la infrecuencia en el tipo de mutación.
Reporte de caso
Se reporta el caso de una mujer de 33 años de edad, médico de profesión, procedente y natural de Villavicencio (Meta, Colombia) quien consulta por sensación de descargas de un cardiodesfibrilador implantable (CDI). Tiene antecedente de taquicardia ventricular polimórfica catecolaminérgica (TVPC), diagnosticada en un estudio electrofisiológico en el año 2010, este diagnóstico se motivó por la presencia de palpitaciones, extrasístoles ventriculares frecuentes y sensación de presíncope.
Contaba con dos pruebas de esfuerzo con criterios de incompetencia cronotrópica, pero sin inducción de arritmias ventriculares, resonancia cardiaca con adelgazamiento de la pared libre hacia el ápex con grosor de la pared de 1 mm por lo que fue difícil evaluar infiltración grasa, prueba de Chagas negativa y estudio electrofisiológico que indujo taquicardia ventricular bidireccional (protocolo con ocho latidos de tren basal a 400 ms con 2 extraestímulos 300 ms y 280 ms, con un latido sinusal seguido de una taquicardia ventricular (TV) bidireccional sostenida, (Figura 1); en dicho estudio no se utilizaron agonistas adrenérgicos. Por la presencia de disfunción sinusal y riesgo de muerte súbita se implantó un CDI bicameral en el contexto de prevención secundaria. Posterior a lo relatado, en el año 2015 presenta agotamiento de la batería por lo que se realiza cambio del generador y, en el año 2018, por descargas inapropiadas secundarias a ruido en electrodo ventricular derecho se abandona el mismo y se realiza implante de nuevo electrodo de alto voltaje. Hasta el año 2022, el dispositivo se encontraba normofuncionante, manteniendo manejo médico farmacológico con bisoprolol. Al examen físico y paraclínicos no se encontraron alteraciones, la telemetría del dispositivo presentaba ruido en el electrodo ventricular que condicionaba descargas inapropiadas. Se explicó a la paciente la necesidad de implantar nuevo electrodo; sin embargo, se negó a una nueva intervención.
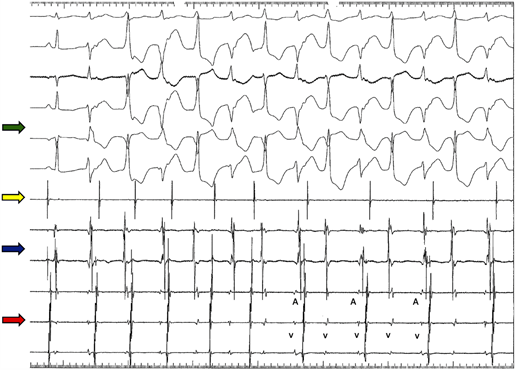
Figura 1 Estudio electrofisiológico. Se observa cambio del eje del QRS latido a latido, taquicardia ventricular bidireccional y seno coronario con clara disociación auriculoventricular. Flecha verde: indica el electrocardiograma de superficie en orden descendente (DI, DII, AVL, AVF, V1 y V6) en donde se observa, inicialmente, un latido sinusal y luego TV bidireccional. Flecha amarilla: electrograma auricular (catéter en aurícula) en donde se evidencia disociación auriculoventricular (AV). Flecha azul: catéter ubicado en región del anillo tricúspideo con disociación AV. Flecha roja catéter ubicado en seno coronario (V: electrograma ventricular y A: electrograma ventricular) con disociación AV.
En la hospitalización actual, el electrocardiograma (EKG) documentó ritmo sinusal alternando con ritmo de estimulación. Dado que la paciente no presentó arritmias ventriculares en más de 13 años de seguimiento, la no autorización de explorar nuevas intervenciones quirúrgicas y el adecuado funcionamiento del dispositivo con respecto a la estimulación; se decidió en conjunto con la junta institucional, apagar las terapias de alto voltaje.
En el seguimiento, en una nueva revisión por sistemas, se identificaron episodios periódicos de parálisis muscular asociados al ejercicio intenso así como clinodactilia, frente prominente, micrognatia e implantación baja de las orejas (Figura 2), se revisaron registros electrocardiográficos previos (2010) donde se documentó ondas U prominentes (Figura 3) por lo que se sospechó SAT.
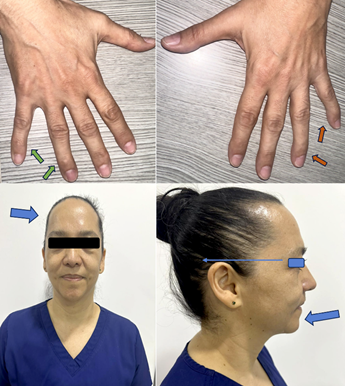
Figura 2 Clinodactilia mano derecha (flecha verde) y mano izquierda (flecha naranja). En la parte inferior de la imagen se identifican otras características físicas (flecha azul) como lo son frente prominente, baja implantación del pabellón auricular y micrognatia.
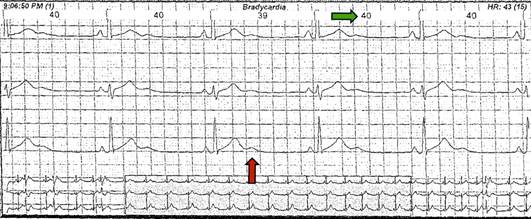
Figura 3 Holter año 2010. Bradicardia sinusal (flecha verde, la paciente se encuentra a 40 latidos por minuto) con ondas U gigantes características (fecha naranja) del SAT.
Actualmente, la paciente a 1 año de seguimiento se encuentra asintomática. El reporte de la prueba genética reportó la variante en heterocigosis del gen KCNJ2 (c.224C>T(p.Thr75Met)), hallazgo compatible con SAT.
Discusión
La muerte súbita arrítmica secundaria a canalopatías sigue teniendo un alto impacto clínico dado el retraso en el diagnóstico 3, se debe tener una alta sospecha y el SAT no es ajeno a este fenómeno. La prevalencia real de esta condición es desconocida, pero se estima que es de 1:1 000 000 personas 4. El diagnóstico del síndrome se propone por la presencia de una triada clínica (Tabla 1) que se apoya en la presencia de una mutación relacionada (la mutación se comprueba en el 70% de los casos que lo clasifica al SAT tipo 1 y cuando se encuentra ausente como tipo 2, la penetrancia de la mutación patológica es variable del 80 a 95%) 5; sin embargo es de aclarar que el diagnóstico de esta entidad es netamente clínico 6. En la descripción de nuestro caso documentamos que la paciente cumple criterios por la presencia de parálisis muscular periódica y arritmia ventricular; si bien la presencia de TV bidireccional es típica de taquicardia ventricular polimórfica catecolaminérgica no es exclusiva de esta condición (Tabla 2) 7, el estrés físico no logró inducir esta arritmia en la paciente y la mutación fue característica de SAT 8.
Tabla 1 Características de la enfermedad
| Definición | Epidemiología | Clínica | Diagnóstico | Tratamiento |
|---|---|---|---|---|
| Es un síndorme arrítmico con un sustrato genético que se caracteriza por la presencia de arritmias ventriculares (TV polimórfica TV bidireccional, Extrasístoles ventriculares), rasgos dismórficos a nivel esquelético y paralisis muscular periodica. Tipo 1 (presencia de mutación documentario) | Se estima una prevalencia de 1:1.000.000 Mutación en el 70% Herencia autosómica dominante Penetrancia variable 80 a 95% | Diferentes presentaciones Asintomáticos Palpitaciones Síncope o presíncope Tormenta arrítmica Muerte súbita Parálisis muscular periódica Cambios físicos (clinodactilia, escoliosis, hipertelorismo, anomalías dentales, etc.) | Criterios clínicos Mutación del gen KCNJ2 o KCNJ5 Niveles de potasio (normales, bajos o altos durante los episodios de debilidad) Hormonas tiroideas normales Estudios de conducción nerviosa normales usualmente Electrocardiograma y holter con arritmia ventricular Ecocardiograma normal | Multidisciplinario Reposición de potasio en los episodios de parálisis muscular Evitar desencadenantes conocidos (estilos de vida) Diuréticos ahorradores de potasio o inhibidores de la anhidrasa carbónica CDI en pacientes con sincope arrítmico o muerte súbita abortada Flecainide Betabloqueador Evitar medicamentos que prolonguen el QT Consejería genética |
Tabla 2 Causas de taquicardia ventricular bidireccional
| Etiología | Posible mecanismo |
|---|---|
| TVPC | Sobrecarga de calcio intracelular |
| Isquemia aguda | Posdespolarizaciones tardías |
| Cardiopatía isquémica | Doble reentrada o una reentrada con doble salida |
| Toxicidad por digoxina | Sobrecarga de calcio intracelular |
| Sarcoidosis | Posdespolarizaciones tardías |
| Síndrome de Andersen-Tawil | Posdespolarizaciones tardías |
| Miocarditis | Posdespolarizaciones tardías |
| Parálisis hipocalémica familiar | Desbalance de las corrientes de potasio |
| Neoplasias cardiacas | Doble reentrada o una reentrada con doble salida |
| Sobredosis de cafeína | Posdespolarizaciones tardías |
| Vasculopatía en aloinjerto coronario | Dos focos parasistólicos |
Adaptado de la referencia (7). TVPC: taquicardia ventricular polimórfica catecolaminérgica.
Además, se resalta la importancia de tener en cuenta que los síntomas pueden ser atemporales y no manifestarse de forma simultánea al momento de la consulta, lo que configura un reto diagnóstico. Es imperativo buscar síntomas neurológicos ante la presencia de arritmias ventriculares de origen no claro o síntomas cardiacos en pacientes con parálisis periódica 9. Se debe examinar al paciente buscando anomalías genéticas menores como clinodactilia, escoliosis, implantación baja de los ojos entre otras. En nuestra paciente se presentó un retraso de más de 10 años en el diagnóstico por no buscar de forma activa la presencia de los síntomas neurológicos, además, se pasó por alto la presencia de anomalías esqueléticas menores.
En todos los pacientes que se sospeche SAT se indica tomar niveles de potasio (basales y durante los episodios de debilidad; los cuales pueden ser normales, bajos o altos), hormonas tiroideas y estudios de conducción nerviosa (usualmente normales). Un estudio de 11 pacientes encontró una respuesta anormal en el posejercicio 10 del 82%. La mutación en el canal de potasio (70%, KCNJ2 o KCNJ5) es la anomalía genética distintiva, en nuestra paciente se encontró una mutación tipo missense con cambio de citocina por timina en el codón 224 del gen KCNJ2, dicha mutación genera una sustitución del aminoácido treonina por metionina en la posición 75 de la proteína que produce unas propiedad fisicoquímicas diferentes. La variante KCNJ2 (NM_000891.3):c.224C>T; p.Thr75Met se encuentra ausente de las bases de datos de población general (https://gnomad.broadinstitute.org/variant/rs104894585) (PM2_Sup).
La significancia de estos hallazgos son patogénicos y se describen nueve casos en la literatura de esta mutación especifica 11-13; no se encuentra reportada en gnomAD, por lo cual se presume una frecuencia alélica (MAF) menor del 1%. Esta variante se ha descrito en múltiples pacientes con síndrome de Andersen-Tawil, con un fenotipo variable, con características dismórficas observadas en todos los pacientes y alta penetrancia de parálisis periódica en varones 12-16. Adicionalmente, esta variante se ha asociado a un alto riesgo de eventos cardiacos en un reporte 11.
El gen KCNJ2 está asociado de manera definitiva con el síndrome de Andersen-Tawil (OMIM#70390) con un modelo de herencia autosómico dominante. Con base en los criterios ACMG la variante se clasifica como patogénica. El tratamiento 17 de estos pacientes es multidisciplinario (Tabla 1) que debe enfocarse en el manejo y prevención de los episodios de debilidad muscular, estratificación del riesgo de muerte súbita, evitar el uso de medicamentos que prolonguen el QT, consejería genética (hijos y familiares) así como seguimiento 18. Actualmente se encuentra en curso un ensayo clínico en donde se comparan estrategias farmacológicas que incluyen beta bloqueador, calcioantagonistas y flecainide con el objetivo de reducir la carga arrítmica 19.
En conclusión, el diagnóstico de esta condición requiere un alto índice de sospecha; se debe ser exhaustivo en el interrogatorio y las pruebas genéticas son esenciales en el abordaje integral diagnóstico de estos pacientes.

