











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by chronic inflammation due to the production of autoantibodies that target almost any organ, leading to a diverse range of clinical manifestations. Cardiovascular (CV) complications are 7.5- to 17.0-fold more common in patients with SLE compared to the general population.1 The CV risk in SLE patients is increased by 50%, which is twice as high as the risk typically reported by the Framingham study.2 Life-threatening conditions such as pericarditis, myocarditis, and venous thromboembolism (VTE) further elevate cardiovascular mortality in these patients.
VTE is a major cause of death and morbidity in SLE. The chronic inflammation associated with the disease contributes to a prothrombotic state, raising the risk of venous thrombosis. Pulmonary thromboembolism (PE) is the most concerning manifestation of VTE, occurring three times more frequently in SLE patients compared to the general population.3 Among these cases, 30-40% are associated with antiphospholipid antibodies (aPL), and 50-70% meet the criteria for systemic antiphospholipid syndrome (APS), which is characterized by multiple venous or arterial thrombosis.4,5 Cases where thromboembolism is the initial cardiovascular manifestation are documented primarily through case reports.
Autoimmune diseases are associated with a higher risk of developing VTE, including PE, mainly due to systemic inflammation that is typical within their pathophysiology. SLE patients show a higher risk of PE, and the incidence is about 7.29%.6 Furthermore, the risk of VTE increases in SLE if aPL and APS are present. In VTE and PE patients, the prevalence of APS has been reported to be 9% in patients aged 18 to 50, 5.8% regardless of age 7 and 30% when selective autoantibodies for APS (aCL, LAC, and/or anti-B2GP1) are used.8 This high prevalence underscores why comprehensive evaluation for systemic autoimmune disease and APS is vital for patient survival, as these conditions represent high-risk factors with poor outcomes.
The management of PE is well established; nevertheless, the concomitant occurrence of cardiac tamponade, which in conjunction is extremely rare, contraindicates the use of fibrinolytics. Managing each condition requires a tailored approach, and restoring hemodynamic stability should guide the final treatment decisions. Herein, we present a rare case of a woman with recurrent PE who experienced cardiac tamponade, which was the initial presentation of SLE associated with APS.
Case report
A 42-year-old woman with a history of diabetes and obesity presented with a sore throat, headaches, diaphoresis, progressive dyspnea, and sudden, intense chest pain that worsened with thoracic movement during breathing (Figure 1). Symptoms had begun a month prior, and there was no improvement with symptomatic medications. Her vital signs were a heart rate of 120 beats per minute, a systolic blood pressure of 85 mmHg, a temperature of 35 °C, and an arterial oxyhemoglobin saturation of 85%. D-Dimer level was 7 µg/mL. Computed tomography pulmonary angiography (CTPA) revealed acute PE in the main branches. The patient was classified as high-risk due to the presence of hemodynamic instability. Although fibrinolytic therapy was initially administered, it was determined as ineffective, and the patient continued to exhibit signs of hemodynamic instability. Consequently, a decision was made to proceed with catheter thrombectomy in conjunction with parenteral anticoagulation using an infusion of unfractionated heparin. The patient was discharged after three days of hemodynamic stability and no complications under DOAC therapy (Rivaroxaban).

Figure 1 Patient clinical evolution timeline. CTPA: Computed tomography pulmonary angiogram. TTE: Transthoracic echocardiography. EKG: Electrocardiogram. mPA: Main pulmonary artery. DOAC: Direct oral anticoagulants. FFP: Fresh frozen plasma. PC: Pericardiocentesis. ICC: Intercostal catheter. SLE: Systemic Lupus Erithematosus. APS: Antiphospholipid syndrome.
Six days post-thrombectomy, the patient presented to the outpatient clinic with progressive dyspnea, nocturnal diaphoresis, a headache, and a non-productive cough. An echocardiogram performed at an external center reported pericardial effusion, and together with clinical features of hypotension, increased jugular venous pressure, and distant heart sound, the suspicion of cardiac tamponade was established.
The patient was referred to our emergency department, arriving with hemodynamic instability, tachycardia, and tachypnea, increased lactate 3 mmol/L, D-dimer 7.45 µg/mL, and an INR >8.4. Admission electrocardiogram (ECG) revealed sinus tachycardia, right axis deviation, low QRS voltage, no ST-segment changes, an incomplete right bundle branch block, and a slightly visible McGinn-White pattern (S1Q3T3). A repeat echocardiogram was performed at our center, confirming the diagnosis of cardiac tamponade. (Figure 2). One unit of fresh frozen plasma was administered, obtaining an INR of 2.6, and pericardiocentesis was performed with the removal of 400 cc serous liquid, which resulted in hemodynamic stabilization, improvement of perfusion and a lactate of 1.4 mmol/L. CTPA report revealed findings consistent with a PE affecting the main pulmonary artery, extending into the right main and segmental branches. Notable improvement in the pericardial effusion was observed. Additionally, the CTPA identified a right pulmonary infarction and bilateral pleural effusions (Figure 3). Consequently, an intercostal catheter was inserted, draining 860 cc of serohematic fluid.
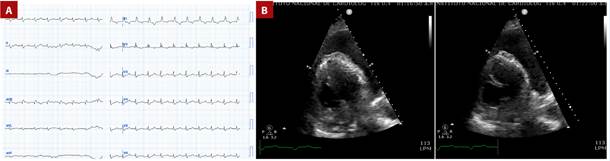
Figure 2 A. Twelve-lead electrocardiogram performed at admission. B. Echocardiogram revealing severe pericardial effusion.
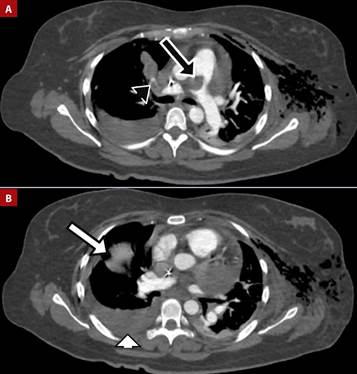
Figure 3 A. Pulmonary embolism in the main pulmonary artery (black arrow) extending into the right main and segmental branches (black arrowhead). B. Pulmonary infarctions (white arrow) and pleural effusion (white arrowhead).
Anticoagulant therapy was initiated with unfractionated heparin and later switched to vitamin K antagonists (VKA) as the patient improved. During the hospital stay, the patient developed ascites and pleuritis, which raised suspicion for polyserositis. The laboratory workup reported hypocomplementemia, leukopenia, positive antinuclear antibodies (ANAs), negative lupus antibodies (IgM anti-β2GP and anti-DNA), positive IgG lupus anticoagulant (LAC), and positive IgG anticardiolipin (aCL), with an SLE SLEDAI score of 3 starting methylprednisolone 1g IV.
During her clinical course, the patient remained hemodynamically stable and asymptomatic, with no evidence of respiratory distress or systemic inflammatory response. She was discharged with VKA, hydroxychloroquine, prednisone 1 mg/kg, and metformin. The follow-up in the clinic revealed an indolent course, with a positive second determination of IgG LAC and IgG aCL antibodies 12 weeks later, which confirmed the diagnosis of APS.
Discussion
Patients with SLE have a higher risk of CV events, with mortality rates tripling healthy people. Thrombotic events, primarily pulmonary, account for 26.5% of these incidents.9 SLE induces a thrombotic state, especially in patients with positive antiphospholipid antibodies. This association has been identified in up to 50% of SLE patients and is responsible for 26.7% of deaths among them.4,10
Focusing on the underlying cause of PE improves outcomes for target management; therefore is essential to determine the etiology/origin. Autoimmune disease is involved in a significant percentage of cases, and positive antiphospholipid antibodies are present in approximately 20% of patients.11 The diagnosis of APS and SLE increases the likelihood of recurrent thrombotic events by 70%(12) and raises the 10-year mortality rate to 44%, with a 5-year mortality rate of 26%.(4,9) This underscores the critical importance of timely diagnosis.
For the diagnosis of SLE, the EULAR/ACR 2019 classification criteria include clinical data such as constitutional, neuropsychiatric, mucocutaneous, serous, musculoskeletal, and renal alterations, as well as immunological data such as the positivity of specific antibodies and hypocomplementemia.13 Sapporo/Sydney criteria for APS diagnosis comprehend thrombotic events or obstetric mortality and the presence of antiphospholipid antibodies: anticardiolipin (aCL), LAC, and/or anti-B2GP1 in two determinations 12 weeks apart.14 Thrombotic events are generally more commonly associated with LAC, and anti-B2GP1, and aCL IgG.15,16 The absence of specific autoantibodies does not exclude APS or SLE; a complete clinical evaluation is crucial for the diagnosis.17
In this case, the patient displayed a risk factor for PE (obesity), nevertheless, recurrent PE, hypocomplementemia, leukopenia, positive ANAs and LAC, and pericardial effusion with evidence of cardiac tamponade indicate that the hemodynamic instability presented may not be solely attributable to the PE and strongly suggest SLE. At the time of intervention, the patient only had one positive immunological test for the APS criteria; despite this, the presence of LAC suggests an autoimmune etiology. The management does not differ from patients without autoimmune disease, and risk classification is required.
To improve outcomes in the management of PE, it is necessary to stratify the predisposing or risk factors that can determine the severity of the event. Our case is classified as high-risk due to hemodynamic instability and its autoimmune origin. ESC guidelines recommend initial management with reperfusion therapy and supportive measures, either pharmacological or mechanical.18 However, our patient's INR >8.4 and cardiac tamponade contraindicate the use of reperfusion therapy. Therefore, the decision was made to address the cardiac tamponade through pericardiocentesis, which resulted in the stabilization of hemodynamic status. Subsequent findings of pleural effusion, suggestive of serositis associated with lupus activity, necessitated an adjustment in the patient's management strategy to include not only anticoagulation but also the consideration of concurrent corticosteroid therapy.16
The simultaneous occurrence of PE and cardiac tamponade is extremely rare and challenging to manage. This is primarily because the treatment for PE, which often involves fibrinolytic therapy, is contraindicated due to the high risk of bleeding associated with cardiac tamponade.19 Treatment decisions are based on a risk-benefit analysis and are always aimed at achieving hemodynamic stability.18 Most case reports attribute this combination to Drug-Induced Lupus Erythematosus.20 A thorough clinical history and a specific diagnostic approach, just like in our case, are crucial for an accurate diagnosis.
In conclusion, the occurrence of recurrent PE with cardiac tamponade as the initial manifestation of SLE and APS is rare and entails considerable difficulties. This unusual combination underscores the critical importance of identifying the underlying cause of PE, especially in such intricate cases. The association between SLE and APS in PE patients is linked to worse outcomes, highlighting the necessity of thorough evaluation and targeted management. Proper risk stratification and appropriate anticoagulation are essential for preventing recurrent thrombotic events and improving patient outcomes.

