











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introducción
La gran variabilidad patológica del cáncer y su creciente resistencia farmacológica están limitando cada vez más la eficacia de muchos tratamientos y empeorando el pronóstico de los pacientes 1. En la búsqueda de nuevas estrategias terapéuticas para superar estas dificultades, diversos estudios se vienen enfocando en las vías metabólicas que están comprendidas en el desarrollo y progresión del cáncer, para así encontrar posibles puntos de bloqueo metabólico y detener su avance. Entre esos posibles blancos se encuentra la proteína fosfatidilinositol-3-quinasas (PI-3K). Esta enzima cumple un papel importante en la regulación del metabolismo, la inmunidad y otras funciones celulares; al mismo tiempo, se conoce que el gen PIK3CA es uno de los más comunes que sufre mutaciones en el cáncer (14 % en todos los cánceres), por lo que ya se han desarrollado fármacos dirigidos contra PI-3K en los últimos años 1-3. Se espera que los paninhibidores de la PI-3K puedan ayudar a superar la resistencia del cáncer a una amplia variedad de terapias como la quimioterapia, la radiación y las terapias dirigidas 4,5.
La PI-3K es una familia de quinasas de lípidos que se clasifican en tres clases: I, II, y III, donde la clase I se subdivide en dos subtipos A y B 6. Son transductores de señales de algunos receptores tirosina quinasas, receptores acoplados a las proteínas G y GTPasas, que regulan, finalmente, diversos procesos celulares como el crecimiento, proliferación, diferenciación, migración, movilidad y apoptosis 1,2. Las PI3-K de la clase IA están conformadas por una subunidad regulatoria p85 (p85α, p85β, p55α, p55γ, p50α) y una catalítica p110 (p110α, p110β, p110γ, p110δ) 6. La subunidad catalítica p110 consiste en un dominio de unión al adaptador (ADP), un dominio de unión a RAS (RBD), un dominio C₂ de unión a membrana (C₂), un dominio helicoidal y un dominio catalítico C-terminal (secciones N y C-terminal), el cual alberga la región bisagra donde se une el adenosín trifosfato (ATP, por sus siglas en inglés) 2. Por otra parte, la subunidad reguladora p85 está compuesta de los dominios SH3, regiones de homología-punto de ruptura (BH), iSH2, cSH2 y nSH2, los cuales deben separarse de la subunidad p110 para poder activar PI-3K 4. Las mutaciones en PI3K que generan una transformación oncogénica están albergadas en la subunidad catalítica p110 y en los dominios RBD, helicoidal (E542K, E545K) y catalítico (H1047R) 7. Las mutaciones somáticas confieren una ganancia de función que conlleva un exceso de actividad por parte de PI-3K 4, y potencia los eventos de activación entre p110 y p85 2. Adicionalmente, se ha observado que cuando se elimina la actividad de PI-3K, el crecimiento del tumor se detiene 8.
Con base en la caracterización de la estructura de PI-3K y de su efecto citostático al ser inhibido, se han desarrollado fármacos para lograr la estabilización del tumor 2. Estos fármacos son generalmente inhibidores competitivos del ATP y pueden clasificarse en paninhibidores, inhibidores específicos e inhibidores duales de PI3K/mTOR 6. Los primeros inhibidores (paninhibidores) desarrollados fueron la wortmanina y el LY294002 9,10; luego, en 2014, el idelalisib (inhibidor de PI3Kδ) se convirtió en el primer inhibidor de PI3K aprobado 2. Hasta la fecha, existen cinco inhibidores de PI-3K aprobados por la FDA: copanlisib, idelalisib, umbralisib, duvelisib y alpelisib 6. A pesar de ello, algunas limitaciones han dificultado la realización de los ensayos clínicos y la aprobación de estos fármacos, como la baja tolerancia a los paninhibidores e inhibidores duales de PI3Kα/PI3Kδ, la resistencia farmacológica intrínseca y adquirida y los bucles de retroalimentación de señalización que neutralizan la inhibición de PI3K 2. Sumado a esta situación, existe también la dificultad para alcanzar la selectividad de isoforma, incluso los actuales inhibidores aprobados pueden afectar a más de una isoforma PI-3K en el entorno clínico 2.
Dentro de los diferentes mecanismos por los que el cáncer desarrolla resistencia a los inhibidores de PI-3K destacan las mutaciones y amplificación de PI-3K, la toxicidad farmacológica, la retroalimentación positiva que conduce a mecanismos compensatorios, el ácido ribonucleico (ARN) no codificante que regula la señalización de PI-3K, el aumento de la producción de insulina y otros mecanismos misceláneos 6. Por esta razón, además de la baja selectividad de isoforma, se ha motivado la búsqueda de nuevos fármacos que puedan ayudar a resolver estos problemas. En este estudio, se utilizan técnicas de reposicionamiento farmacológico in silico para encontrar la actividad inhibitoria de PI-3K en fármacos aprobados por la FDA que originalmente no fueron diseñados para esta indicación 11. Se ha empleado una aproximación computacional para identificar rápidamente los fármacos que podrían inhibir competitivamente a PI-3K, utilizando nueve modelos de farmacóforos diferentes. Esta aproximación ofrece un método de bajo costo y rápido para sugerir fármacos, además de contar con información de su seguridad, que ha sido ampliamente estudiada en estudios preclínicos y clínicos necesarios para su aprobación 11.
Materiales y métodos
Diseño y población de estudio
El presente estudio es de tipo descriptivo y se llevó a cabo mediante ensayos in silico en los laboratorios de la Universidad de Piura. Los estudios in silico utilizaron aproximaciones y herramientas bioinformáticas especializadas, y se consideró como población el dominio catalítico de PI-3K. Para obtener la estructura de PI3Kα (p110), primero se usó la base de datos UniProt (código: P42336) como referencia, ya que las estructuras que están cristalizadas en RCSB-PDB (www.rcsb.org) estaban incompletas. El modelo final del dominio catalítico de PI3Kα se obtuvo de Alpha Fold (https://alphafold.ebi. ac.uk/), el cual tiene un nivel de confianza alto.
En la literatura se identificaron los inhibidores de PI3Kα y, posteriormente, se encontró su estructura cocristalizada con estos en el RCSB-PDB (inavolisib: 8EXV, taselisib: 8EXL, CH5132799: 3APC, alpesilib: 7MYO, ZSTK474: 2WXL).
Luego, se alineó cada estructura con el modelo de Alpha Fold en PyMOL. Se eliminó el PI3Kα cocristalizado, y solo se conservó la estructura del inhibidor en complejo con el modelo de Alpha Fold. El mismo proceso se realizó con la estructura cocristalizada que contiene ATP (PDB: 1E8X).
Variables y mediciones
Los ensayos de aproximación in silico consideraron varias variables: los fármacos aprobados por la FDA obtenidos mediante cribado virtual, las energías de unión en kcal/mol derivadas del acoplamiento molecular entre el dominio catalítico de PI-3K y los fármacos, así como los aminoácidos involucrados en la interacción fármaco-proteína.
Para la realizar el cribado virtual, se fabricaron farmacóforos usando el servidor web Pharmit (http://pharmit.csb.pitt.edu/). Cada farmacóforo se construyó con cinco posiciones claves. Estas posiciones fueron decididas en base a las interacciones inhibidor-proteína (inavolisib, taselisib, alpesilib, CH5132799 y ZSTK474) más importantes, obtenidas en el servidor web PLIP (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index).
Se complementó con la búsqueda en la literatura acerca de las regiones y los aminoácidos claves en el proceso de fosforilación entre ATP y PI3Kα. Con esta información se definieron las posiciones claves y se obtuvieron sus coordenadas XYZ en Pharmit y PyMOL. El cribado virtual se realizó utilizando la biblioteca de medicamentos aprobados por la FDA. Se consideró una tolerancia de 0,5 en la opción "Receptor-Exclusive shape'' para restringir el tamaño de la molécula al bolsillo de unión del ATP.
Las energías de unión se obtuvieron usando el programa YASARA, mediante la opción "BindEnergy". Para este procedimiento, primero se limpiaron las estructuras utilizando la opción "Clean" y se añadieron los hidrógenos faltantes con la opción "AddHydr". Luego, alrededor del sistema se creó una caja de 10 Å para cubrir toda la proteína y, finalmente, se llevo a cabo una minimización de energía seguida del acomplamiento. Los aminoácidos de las interacciones fármaco-proteína se obtuvieron del servidor web PLIP.
Análisis estadístico
Se usó la estadística descriptiva para evaluar la afinidad de la unión entre los fármacos seleccionados y el dominio catalítico de PI-3K. Los valores se expresaron en un gráfico de barras para visualizar la distribución de las energías de unión. Además, se calculó la frecuencia de interacciones específicas en los fármacos seleccionados como candidatos al reposicionamiento farmacológico.
Consideraciones éticas
Este estudio fue aprobado por el Comité Institucional de Ética en Investigación de la Universidad de Piura (Nro: PREMED0820219). Al ser un estudio con abordaje in silico, no se requirió la intervención de seres humanos o el uso de muestras biológicas. Se desarrolló en los laboratorios de investigación de Cultivo Celular, Inmunología y Biología Celular y Análisis de Proteína y Bioinformática de la Universidad de Piura.
Resultados
En base a las interacciones y la literatura, en este estudio se propone nueve modelos de farmacóforos (Tabla 1), cada uno con cinco características, donde se reflejan las posiciones más importantes usadas por los inhibidores existentes y las regiones de unión delATP en las quinasas 12. Los farmacóforos 1, 2 y 3 contienen posiciones que se ubican en la región de adenina y la región enterrada. Los farmacóforos 4 y 5 agregan posiciones en la región accesible al solvente, los 6 y 7 agregan posiciones en la región del azúcar, mientras que 8 y 9 lo hacen en la región de unión al fosfato.
Tabla 1 Modelos de farmacóforos y sus distintas características
| Modelo | Características |
|---|---|
| Farmacóforo 1 | HBA1, Hyd1, HBA2, Hyd2, HBA3 |
| Farmacóforo 2 | HBA1, Hyd1, HBA4, Hyd2, HBA3 |
| Farmacóforo 3 | HBA1, Hyd1, Hyd3, HBA5, HBA6 |
| Farmacóforo 4 | HBA1, Hyd1, HBDo1, Hyd2, HBA3 |
| Farmacóforo 5 | HBA1, Hyd1, Hyd2, HBA3, HBA7 |
| Farmacóforo 6 | HBA1, Hyd1, Hyd2, HBA3, HBDo2 |
| Farmacóforo 7 | HBA1, Hyd1, Hyd2, HBA3, HBA8 |
| Farmacóforo 8 | HBA1, Hyd1, Hyd2, HBA3, HBA9 |
| Farmacóforo 9 | HBA1, Hyd1, Hyd2, HBA3, HBA10 |
HBDo: Enlace de hidrógeno donador; HBA: enlace de hidrógeno aceptor; Hyd: hidrofóbico; Aro: aromático.
Los modelos de farmacóforos generados fueron usados para buscar fármacos que contengan estas características en la biblioteca de medicamentos aprobados por la FDA, disponible en Pharmit. Inicialmente, se obtuvo un total de 33 fármacos en los nueve modelos propuestos (Tabla 2).
Luego de identificar aquellos que se encontraban repetidos en más de un modelo de farmacóforo, se procedió a elegir solo uno, según su energía de unión, y eliminar el resto. Al finalizar este proceso, se mantuvieron 22 fármacos.
Tabla 2 Fármacos obtenidos en cada modelo
| Modelo | Fármacos aprobados por la FDA |
|---|---|
| 1 | Sulfadoxina, cefonicida, apalutamida |
| 2 | Entecavir |
| 3 | Cefamandol, dexametasona, sulfametazina, terazosina, trazodona |
| 4 | Azilsartán, larotrectinib, regadenosón, sildenafil |
| 5 | Ceftazidima, entecavir, omeprazol, regadenosón, ritodrina, vardenafilo |
| 6 | Nelarabina, riboflavina |
| 7 | Azelastina, bosentán, entecavir, fostamatinib, riboflavina, tenofovir |
| 8 | Fostamatinib, pralatrexato |
| 9 | Entecavir, nizatidina, pralatrexato, tedizolid |
Para cada fármaco obtenido se midió la energía de unión (kcal/mol) del complejo formado por PI3-K y los fármacos aprobados por la FDA. Lo mismo se hizo para los inhibidores existentes de PI-3K (inavolisib, taselisib, CH5132799, alpelisib, ZSTK474) con PI-3K (Figura 1A).
Dentro de los fármacos aprobados por la FDA, pralatrexato (57,2 kcal/mol) y entecavir (56,1 kcal/mol) fueron los que tenían mayor energía de unión a PI-3K, incluso llegando a superar la mayoría de los inhibidores de PI-3K (alpelisib: 53,28 kcal/mol, inavolisib: 53,13 kcal/mol, CH5132799: 43,31 kcal/mol, ZSTK474: 47,02 kcal/mol). Dentro de los inhibidores existentes de PI3Kα, taselisib (59,75 kcal/mol) fue el que tenía el mayor valor de energía de unión y, además, era superior a todos los otros fármacos.
Se restaron los valores de energía de unión individuales de los fármacos aprobados por la FDA, que se obtuvieron en el cribado virtual con el promedio de los inhibidores específicos ya existentes para PI-3K. Con esto se pudo identificar diez posibles candidatos que tenían una energía de unión dentro del rango de -10 y +10 (Figura 1B). En la Tabla 3, se describen las características de los enlaces que estos diez fármacos candidatos al reposicionamiento tienen con el sitio activo de PI3Kα.

Figura 1 Energía de unión entre PI3K y fármaco. A. Las barras negras representan la energía de unión de PI-3K con los fármacos aprobados por la FDA para otros usos, mientras que las barras blancas representan la energía de unión de PI-3K con sus inhibidores específicos desarrollados. B. Diagrama de dispersión de la energía de activación entre PI3Kα y fármacos. Se consideró como aceptable la energía de unión de los fármacos que se encontraban entre el rango de -10 a +10 (líneas rojas). En sombreado gris, se aprecia los fármacos inhibidores específicos de PI3Kα.
Tabla 3 Características de los fármacos candidatos al reposicionamiento
| Fármacos | Energía de unión (kcal/mol) | Puentes de hidrógeno | Interacciones hidrofóbicas | Pi-stacking | Puentes de sal |
|---|---|---|---|---|---|
| Fostamatinib | 48,7 | Arg770 | - | - | Arg770 |
| His855 | |||||
| Gln859 | |||||
| Asp933 | |||||
| Entecavir | 56,1 | Ser773 | Ile800 | - | - |
| Ser774 | Ile932 | ||||
| Val851 | |||||
| Gln859 | |||||
| Ser919 | |||||
| Asp933 | |||||
| Pralatrexato | 56,4 | Ser774 | Ile800 | - | - |
| Lys802 | Tyr836 | ||||
| Asn853 | Val850 | ||||
| Ser854 | Val851 | ||||
| Gln859 | Ile932 | ||||
| Tedizolid | 43,6 | Asp810 | Ile800 | Trp780 | Asp810 |
| Gln859 | Ile848 | Asp933 | |||
| Asp933 | Ile932 | ||||
| Tenofovir | 43,5 | Asp810 | Trp780 | Trp780 | - |
| Gln859 | Ile800 | ||||
| Asp933 | Val850 | ||||
| Gln859 | - | ||||
| Vardenafilo | 45 | Arg770 | Trp780 | Trp780 | |
| Val851 | Ile800 | ||||
| Ser854 | Ile932 | ||||
| Gln859 | - | - | |||
| Regadenosón | 46,2 | Val851 | |||
| Ser854 | |||||
| Gln859 | |||||
| Asp933 | - | ||||
| Apalutamida | 49 | Val851 | Trp780 | - | |
| Ser854 | Ile848 | ||||
| His855 | Val851 | ||||
| Gln859 | Phe930 | ||||
| Asp933 | Ile932 | - | |||
| Cefonicida | 50,6 | Val851 | Ile800 | - | |
| Asn853 | Tyr836 | ||||
| Ser854 | Ile848 | ||||
| His855 | Ile932 | ||||
| Gln859 | |||||
| Azilsartán | 42,9 | Arg770 | Ile800 | - | Lys802 |
| Gln859 | Lys802 | ||||
| Asp933 | Tyr836 | ||||
| Ile848 | |||||
| Val851 | |||||
| Thr856 | |||||
| Ile932 |
Discusión
De los diez fármacos antes mencionados, se han seleccionado tres en particular: fostamatinib, pralatrexato y entecavir, para su posible reposicionamiento farmacológico, tomando como base sus interacciones in silico y estudios de sus posibles usos en cáncer.
El fostamatinib es un inhibidor de la tirosina quinasa esplénica (spleen tyrosine kinase, Syk), que ha sido aprobado para el tratamiento de la trombocitopenia autoinmune crónica en los pacientes en los que ha fallado la terapia convencional 13, aunque su uso viene siendo evaluado también en otras patologías 14. Estudios previos han demostrado que la proteína Syk es un mediador molecular clave en la regulación de la plasticidad epitelial-mesenquimal y en la transición epitelial-mesenquimal (EMT, por sus siglas en inglés). Este proceso es fundamental para el desarrollo de la metástasis a partir de un tumor primario 15. Si bien inicialmente se identificó que los tejidos de pacientes con cáncer presentaban menor expresión de Syk, lo que llevaría a ubicarlo como un posible supresor de tumores 16, estudios experimentales han encontrado que tendría una participación directa en la EMT y sugieren que su inhibición ―usando fostamatinib, por ejemplo― podría disminuir la probabilidad de metástasis 15,17. A nivel de ensayos clínicos, los resultados de fostamatinib aún no son concluyentes, ya que muestran una tolerancia variable y una baja efectividad en algunos tumores sólidos 14,18. La interacción del fostamatinib con quinasas distintas a Syk, como Flt3, JAK, c-Kit, Lck o RET, ya se ha identificado 14, aunque su uso como un posible inhibidor de PI-3K aún no se ha evaluado. En nuestro hallazgo in silico, la interacción del fostamatinib con PI-3K se caracteriza por formar puentes de hidrógeno con His855, Gln859, Asp933 y Arg770, tal como se aprecia en la Figura 2A. Estas interacciones también están presentes en los inhibidores ya existentes de PI3Kα y el ATP. Los enlaces con los residuos de His855 y Gln859 podrían ayudar a tener una mayor especificidad respecto a PI3Kα 19. Se ha visto que la carboxiamida de alpelisib, taselisib e inavolisib también se une a la Gln859 2, lo que podría sugerir que este residuo cumple un rol importante en diferenciar la afinidad entre PI3Kα y las otras isoformas 19. El Asp933 pertenece al motivo DFG (aspartato, fenilalalina y glicina), que está conservado en todas las proteínas quinasas y desempeña una función crítica en la catálisis. Este motivo se une al ion Mg2+ para orientar al fosfato γ del ATP durante su transferencia 20. Cabe resaltar que este residuo también es un blanco frecuente de unión para los inhibidores de PI3Kα anteriormente mencionados 2.
Por otro lado, el pralatrexato es un fármaco que se usa para tratar formas avanzadas o recurrentes de linfoma de células T periféricas. Se trata de un antimetabolito inhibidor análogo del ácido fólico, por lo que impide la replicación del ADN y el ciclo celular. Algunos estudios preclínicos han mostrado un efecto alentador del pralatrexato en el neuroblastoma de alto riesgo (incluso superior al metotrexato) 21, y superior a otros antimetabolitos en el cáncer de pulmón de células no pequeñas (CPCNP) recurrente o refractario y algunos otros tipos de linfomas 22. Inclusive, se ha sugerido que se considere al pralatrexato como un fármaco para reposicionamiento en el tratamiento de la COVID-19, dado su efecto viral añadido a su rol antineoplásico 23. No se ha encontrado literatura que describa el efecto del pralatrexato en la quinasa PI3Kα ni en cánceres con alto índice de mutación de esta enzima, por lo que nuestro hallazgo podría aportar nuevas luces en su uso futuro. En la Figura 2B se muestra la interacción de pralatrexato y PI3Kα, donde se destaca, además de la Gln859, su enlace con la Lys802 catalítica y la Ser854. Se ha descrito que la Lys802 participa en la reacción de transferencia del fosfato en p110α 20,24,25 y es blanco de interacción en el inhibidor específico de PI3Kα, CH5132799 2. Por otra parte, se ha reportado que existen enlaces entre la Ser854 y alpelisib, taselisib e inavolisib 2, lo que sugiere que también es un importante residuo en la función catalítica de PI3Kα.
El entecavir es un antiviral de tipo nucleosídico utilizado desde hace casi dos décadas para el tratamiento de la infección crónica por el virus de la hepatitis B, cuyo efecto biológico es la inhibición de la replicación viral 26. Al ser un análogo de la guanina, su papel en la terapia contra el cáncer ha sido evaluado en aproximaciones de reposicionamiento farmacológico in silico, en donde se sugiere su posible rol como quimioterápico en cáncer de mama, ovario y próstata, y en tejidos con alta actividad de PARP-1 27,28. Sin embargo, no se evalúa su posible rol inhibidor de quinasas clave en la progresión del cáncer como PI3Kα, cuya interacción es graficada en la Figura 2C. De igual forma que con el fostamatinib, presenta la unión con la Gln859 y el Asp933, lo que le conferiría las características antes descritas. Además, se indica que interactúa con la Val851, que se encuentra en la región bisagra de la quinasa. Este mismo residuo es blanco en otros inhibidores conocidos para PI3Kα, como alpelisib, taselisib, inavolisib y CH5132799 2.
Finalmente, cabe resaltar que otros fármacos con buenos resultados in silico contra PI3K ya están siendo evaluados en la terapia contra el cáncer. Un ejemplo es el tenofovir, un medicamento utilizado contra la infección por el VIH, que ha demostrado efectos en algunos tipos de cáncer recurrente o avanzado, como el carcinoma hepatocelular 29,30.
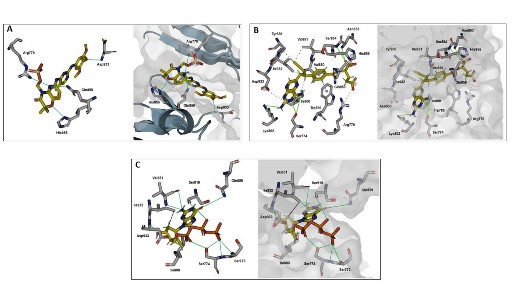
Figura 2 Interacción fármaco-proteína. A. Interacción entre fostamatinib y PI3K. B. Interacción entre pralaxetrato y PI3K. C. Interacción entre entecavir y PI3K. Todas las interacciones se muestran en formato stick (izquierda del recuadro) y formato surface (derecha del recuadro). Los puentes de hidrógenos están representados con líneas de color verde.
En conclusión, este estudio identificó diez fármacos con probabilidad de reposicionamiento farmacológico en el tratamiento del cáncer mediante su interacción con PI3Kα. De estos, fostamatinib, pralatrexato y entecavir fueron seleccionados debido a sus interacciones in silico y estudios de sus posibles usos en cáncer. Estos fármacos están en fase de pruebas experimentales o ensayos clínicos, donde su blanco de interacción son las proteínas quinasas; por lo tanto, la posibilidad de que interactúen con PI3Kα es viable. Además, este proyecto generó nueve farmacóforos que pueden usarse en diferentes bases de datos especializadas para la búsqueda de nuevas moléculas y/o fármacos con capacidad potencial para el reposicionamiento farmacológico. Por consiguiente, se sugiere continuar con la exploración in silico e in vitro, enfocándose en evaluar el rol de estos fármacos en la inhibición de PI3Kα y en los tipos de neoplasias en los que la actividad de esta quinasa o sus mutaciones jueguen un papel clave.

