











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las enfermedades neurodegenerativas, como el Alzheimer y el Parkinson son un gran problema para la salud mundial. Estas enfermedades se caracterizan por una pérdida neuronal progresiva que conduce a un deterioro cognitivo irreversible, deterioro motor y, en última instancia, la muerte. La falta de tratamientos eficaces para estas enfermedades, urgente la necesidad de estrategias terapéuticas innovadoras.1,2,3
La acetilcolinesterasa (AChE), una enzima clave en el sistema nervioso central, desempeña un papel fundamental en la patogénesis de las enfermedades neurodegenerativas. La AChE es responsable de hidrolizar la acetilcolina, un neurotransmisor crucial para la función cognitiva y la memoria. La desregulación de la actividad de la AChE, que a menudo conduce a un exceso de la enzima, contribuye al agotamiento de los niveles de acetilcolina en el cerebro, lo que contribuye a los déficits cognitivos y motores observados en las enfermedades como el Alzheimer y el Parkinson. 1,4
Los inhibidores de la AChE han surgido como una clase prometedora de agentes terapéuticos para enfermedades neurodegenerativas. La inhibición de la actividad de la AChE, tiene como objetivo aumentar los niveles de acetilcolina en el cerebro, mitigando potencialmente los síntomas y ralentizando la progresión de estas enfermedades debilitantes. La búsqueda de nuevos y efectivos inhibidores de la AChE sigue siendo un área activa de investigación, en la identificación y caracterización de candidatos potenciales.1,2,3
La taspina, un alcaloide natural, que ha surgido como un candidato prometedor para la inhibición de la AChE. Estudios previos han indicado el potencial de la taspina para modular la actividad de la AChE, ofreciendo una posible vía terapéutica para el tratamiento de enfermedades neurodegenerativas. Sin embargo, una comprensión integral de las interacciones de la taspina con la AChE sigue siendo difícil de alcanzar, lo que resalta la necesidad de una mayor investigación. 1,2,4,5
En el contexto del descubrimiento de fármacos, los cálculos DFT desempeñan un papel crucial en la comprensión de la reactividad y el comportamiento electrónico de los posibles candidatos a fármacos. Al analizar la estructura electrónica de una molécula, los cálculos DFT pueden proporcionar información valiosa sobre su potencial para interactuar con una proteína objetivo, lo que en última instancia influye en su actividad farmacológica. 4,5,6
Adicionalmente, el acoplamiento molecular es particularmente valioso en las primeras etapas del descubrimiento de fármacos, ya que proporciona un método rentable y eficiente para la selección de un gran número de posibles candidatos a fármacos. Al identificar candidatos prometedores con altas afinidades de unión e interacciones favorables, el acoplamiento molecular puede guiar la selección de compuestos para una evaluación experimental adicional.5,6
El uso de la dinámica molecular se ha consolidado como una metodología de gran importancia. Esta técnica nos ofrece información del comportamiento dinámico de las moléculas en función del tiempo, permitiendo explorar la estabilidad conformacional de las proteínas, dinámicas en las interacciones proteína-ligando, cambios estructurales inducidos e interacciones no covalentes que influyen en los complejos biomoleculares en entornos realistas5. El presente estudio nos permitirá estudiar los cambios conformacionales inducidos en la AChE-Taspina, observando como el ligando se comporta en el sitio de unión a lo largo del tiempo en un entorno acuoso.
Para profundizar en el potencial de la taspina como inhibidor de la AChE, este estudio emplea un enfoque de química computacional, utilizando una combinación de teoría funcional de la densidad (DFT), acoplamiento molecular y simulaciones de dinámica molecular. Este enfoque permite una investigación exhaustiva de las propiedades de la taspina y sus interacciones con la AChE a nivel molecular. Los cálculos DFT brindan información sobre la estructura electrónica y la reactividad de la taspina, el acoplamiento molecular predice la afinidad de unión e identificar interacciones potenciales, mientras que las simulaciones de dinámica molecular simulan el comportamiento dinámico del complejo taspina-AChE a lo largo del tiempo.
PARTE EXPERIMENTAL
Cálculos de química Cuántica
Los cálculos se realizaron con el software ORCA en su versión Orca-5.0.37. Esto se llevó a cabo utilizando el método DFT8. Inicialmente, la taspina fue preoptimizada con wB97M y el conjunto base def2-ZVP; luego, se optimizó su geometría con el funcional wB97M-D4 [9] y el conjunto base def2-TZVPP 10, todo en fase gaseosa. Los cálculos de los índices de Fukui también se realizaron con Orca-5.0.3, utilizando las coordenadas obtenidas en la optimización de la geometría. Para esto se empleó también el funcional wB97M-D4 y el conjunto base def2-TZVPP. Los inputs fueron las moléculas de taspina aniónica, catiónica y neutra. Para determinar el gradiente reducido de la densidad (RDG), se utilizaron dos softwares adicionales: VMD y Multiwfn, que es un programa de análisis de funciones de onda11. Todos los cálculos mencionados anteriormente se realizaron usando una computadora personal AMD Ryzen 7 5800X3D, 3.40 GHz, 8 núcleos, 16 hilos de ejecución.
Acoplamiento molecular
Un paso previo antes de realizar el anclaje molecular fue la búsqueda de la enzima que interactúa con la taspina mediante el uso de SwissTargetPrediction ya que permitió identificar a las posibles proteínas dianas involucradas en su mecanismo de acción. La estructura elegida fue la acetilcolinesterasa (AChE-S) que se descargó del banco de proteínas (RCSB PDB) identificada con el código 4M0E, en la cual se simuló el acoplamiento molecular contra la taspina. La proteína se preparó utilizando el programa UCSF Chimera, donde se removieron las moléculas de agua. Luego, se identificaron los sitios activos de la AChE donde se simularon las interacciones del ligando y los aminoácidos terminales, empleando el servidor en línea CASTp que localizó la cavidad con los sitios activos. Seguido se utilizó la interfaz gráfica AutoDockTools para crear el archivo confing.txt, que contuvo el centro de la caja con las coordenadas en X= 1.154, Y= -41.283 y Z= 13.081, de la cavidad para el acoplamiento. luego, se creó una carpeta de trabajo donde se guardó el archivo config.txt, donde se modificó la exhaustividad a 32 y el número de conformaciones a 1, la taspina y la proteína AChE en formato pdbqt, y un script llamado docking.sh utilizado para realizar el anclaje molecular de forma repetitiva unas 50 veces. Una vez hecha la carpeta de trabajo se empleó el programa AutoDock Vina, desde la terminal de linux ejecutando los comandos chmod +x docking.sh y ./docking.sh. Por último, se realizó una simulación computacional más costosa y exhaustiva mediante el anclaje molecular flexible que nos permite seleccionar una cantidad limitada de los aminoácidos terminales de la cavidad y hacerlos flexibles para que cambien su conformación junto con el ligando durante la simulación.
Método computacional de Simulación de Dinámica Molecular (DM)
Las simulaciones de DM all-atom fueron realizadas considerando las coordenadas atómicas entre la proteína AChE-S y taspina usando Gromacs v5.1.4 con campo de fuerza AMBER99SB y modelo de agua TIP4P. La caja de simulación fue definida con una distancia de 10A° desde el centro del complejo proteína ligando. El sistema preparado fue solvatado con moléculas de agua y neutralizado con la adición de 0.15M de iones Na+ y Cl- 13. El proceso de minimización de energía involucró 50000 pasos por cada descenso seguido por gradientes conjugados. Las condiciones PBC fueron definidas por direcciones x, y, z14 y la simulación fue realizada a temperatura fisiológica de 300K. Se aplicó el algoritmo VERLET para restringir todos los enlaces involucrados con hidrógeno y fuerzas electrostáticas de largo alcance tratadas con PME (Particle mesh Ewald). El sistema fue equilibrado en dos pasos, NVT y NPT a 300K por periodos de 1000ps. Durante la simulación, se utilizaron el termostato V-rescale y Parrinello-Rahman15 para mantener la presión y la temperatura. Se utilizó el algoritmo LINCS para restringir los enlaces y ángulos. Las interacciones de Van der Waals fueron restringidas con el potencial LJ con un límite de 1.0 nm.
RESULTADOS Y DISCUSIÓN
Método de gradiente reducido de la densidad (RDG)
La gradiente reducida de la densidad (RDG) es una herramienta clave en la visualización de interacciones no covalentes [12]. El análisis del RDG revela diversas interacciones atractivas, de Van der Waals, y repulsivas que influyen en la estabilidad y comportamiento de la molécula. El gradiente reducido de densidad (s), un campo escalar de la densidad electrónica, se describe como:
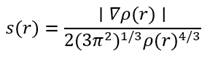
La taspina, al ser una molécula con cuatro anillos aromáticos fusionados, así como grupos éster, éter y una amina primaria, exhibe una red compleja de interacciones intra e intermoleculares. Estas interacciones pueden visualizarse mediante el índice RDG al analizar los valores del parámetro λ2ρ, donde la densidad electrónica y su gradiente indican la naturaleza de las interacciones. Donde H-B (atracciones fuertes: tipo enlace puente de hidrógeno), VdW (Interacciones de Van der Waals) y E.E. (Repulsiones fuertes efectos estéricos).
En la figura 1(a), no se observa una isosuperficie de color azul, lo que sugiere la ausencia de enlaces por puente de hidrógeno. Esto concuerda con la estructura química de la taspina, la cual no posee átomos dadores de puentes de hidrógeno, pero sus átomos de nitrógeno y oxígeno pueden ser aceptores de puentes de hidrógeno. Sin embargo, se observa otras interacciones, como las de tipo Van der Waals (VdW) y repulsiones estéricas (E.E.), estas se representan mediante isosuperficies de color verde y rojo, respectivamente.
Por otro lado, en la figura 1(b), se observa que los valores asociados a las interacciones fuertes (H-B) se encuentran entre -0.35 < sign( 𝜆 2 )𝜌 < -0.25 a.u. Mientras que las interacciones VdW están en el rango de -0.20 < 𝑠𝑖𝑔𝑛( 𝜆 2 )𝜌 < 0 a.u., lo que indica la presencia de interacciones Van der Waals en la región ecuatorial de la molécula principalmente. Finalmente, las isosuperficies de color rojo que representan las repulsiones estéricas se localizan principalmente en el centro de los cuatro anillos aromáticos de la taspina, los valores de estas interacciones oscilan entre 0.10 < 𝑠𝑖𝑔𝑛( 𝜆 2 )𝜌 < 0.20 a.u.
Descriptores Globales y Locales
En la tabla 1 se observan los descriptores globales, estos indican la reactividad general del sistema. La diferencia de energía entre el HOMO y el LUMO da un ΔE = 0.0304 a.u., lo cual sugiere una moderada estabilidad frente a la transferencia electrónica. El valor negativo de la dureza global (η) indica que la molécula es flexible frente a cambios electrónicos, lo que aumenta su reactividad; el valor de la blandura global (s) es el inverso de la dureza, su valor negativo confirma lo mencionado. El valor negativo de la electronegatividad (χ) sugiere que la molécula tiene una baja tendencia a atraer electrones. Por último, el valor de electrofilicidad global (Ψ) sugiere que la taspina tiene baja propensión a aceptar electrones, indicando que se comporta más como nucleófilo que como electrófilo.
Tabla 1 Descriptores globales.
| HOMO | 0.0209 |
| LUMO | -0.0095 |
| ΔE | 0.0304 |
| Energía de ionización (EI) | -0.0209 |
| Afinidad electrónica (AE) | 0.0095 |
| Dureza global (η) | -0.0152 |
| Blandura global (s) | -0.0076 |
| Electronegatividad (χ) | -0.0057 |
| Potencial químico (µ) | 0.0057 |
| Electrofilicidad global (Ψ) | -0.00106875 |
En el análisis de la función de Fukui, al considerar los estados de carga neutro, catiónico (+1) y aniónico (-1), se obtienen los valores de la función de Fukui condensada para ataques nucleofílicos (𝑓−), electrofílicos (𝑓+) y radicalarios (𝑓0), los cuales proporcionan información valiosa sobre los sitios más reactivos de una molécula. Los valores positivos de 𝑓+ y 𝑓− revelan tendencias significativas en los centros carbonados, oxígenos y nitrógenos, indicando su propensión a sufrir ataques nucleofílicos y electrófilos, respectivamente. En la tabla 2, se presentan los descriptores locales calculados para identificar sitios reactivos dentro de la molécula. Los carbonos C6 y C4 presentan valores elevados de 𝑓− de 0.097 y 0.068, respectivamente, lo que indica que estas posiciones son favorables para recibir ataques electrófilos. Por otro lado, los átomos de oxígeno O15 y O7 presentan altos valores de 𝑓+ de 0.065 y 0.054, lo que sugiere que estas posiciones son susceptibles a sufrir ataques nucleofílicos.
Tabla 2 Descriptores locales.
| Átomo | f- | f0 | f+ | Descriptor | Dureza |
|---|---|---|---|---|---|
| catión | neutro | anión | dual | local (au) | |
| C1 | 0.034019 | 0.042151 | 0.050283 | 0.016264 | -0.00024721 |
| C10 | 0.006840 | 0.018218 | 0.029597 | 0.022757 | -0.00034591 |
| C12 | 0.010360 | 0.023776 | 0.037192 | 0.026832 | -0.00040785 |
| C14 | 0.031724 | 0.024874 | 0.018024 | -0.013700 | 0.00020824 |
| C17 | 0.012931 | 0.012469 | 0.012008 | -0.000923 | 1.403E-05 |
| C2 | 0.038276 | 0.037847 | 0.037419 | -0.000857 | 1.3026E-05 |
| C21 | 0.012410 | 0.008328 | 0.004245 | -0.008165 | 0.00012411 |
| C24 | 0.001873 | 0.002124 | 0.002374 | 0.000501 | -7.6152E-06 |
| C28 | 0.006724 | 0.005907 | 0.005090 | -0.001634 | 2.4837E-05 |
| C3 | 0.037324 | 0.044811 | 0.052297 | 0.014973 | -0.00022759 |
| C32 | 0.004645 | 0.003897 | 0.003149 | -0.001496 | 2.2739E-05 |
| C36 | 0.020237 | 0.032738 | 0.045240 | 0.025003 | -0.00038005 |
| C37 | 0.040758 | 0.048573 | 0.056388 | 0.015630 | -0.00023758 |
| C39 | 0.032841 | 0.031661 | 0.030481 | -0.002360 | 3.5872E-05 |
| C4 | 0.068818 | 0.047036 | 0.025255 | -0.043563 | 0.00066216 |
| C42 | 0.007945 | 0.009551 | 0.011157 | 0.003212 | -4.8822E-05 |
| C5 | 0.022658 | 0.035107 | 0.047556 | 0.024898 | -0.00037845 |
| C6 | 0.097263 | 0.056725 | 0.016186 | -0.081077 | 0.00123237 |
| C8 | 0.016054 | 0.023373 | 0.030692 | 0.014638 | -0.0002225 |
| C9 | 0.005421 | 0.024973 | 0.044525 | 0.039104 | -0.00059438 |
| H0 | 0.024424 | 0.026212 | 0.028000 | 0.003576 | -5.4355E-05 |
| H18 | 0.011338 | 0.010353 | 0.009367 | -0.001971 | 2.9959E-05 |
| H19 | 0.022270 | 0.021062 | 0.019854 | -0.002416 | 3.6723E-05 |
| H20 | 0.009366 | 0.008993 | 0.008621 | -0.000745 | 1.1324E-05 |
| H22 | 0.020474 | 0.014904 | 0.009334 | -0.011140 | 0.00016933 |
| H23 | 0.020734 | 0.014836 | 0.008937 | -0.011797 | 0.00017931 |
| H25 | 0.004358 | 0.004268 | 0.004178 | -0.000180 | 2.736E-06 |
| H26 | 0.005620 | 0.005263 | 0.004906 | -0.000714 | 1.0853E-05 |
| H29 | 0.004902 | 0.004240 | 0.003577 | -0.001325 | 2.014E-05 |
| H30 | 0.011857 | 0.011046 | 0.010234 | -0.001623 | 2.467E-05 |
| H31 | 0.008607 | 0.007625 | 0.006644 | -0.001963 | 2.9838E-05 |
| H33 | 0.000103 | -0.000810 | -0.001724 | -0.001827 | 2.777E-05 |
| H34 | 0.007395 | 0.006449 | 0.005502 | -0.001893 | 2.8774E-05 |
| H35 | 0.012650 | 0.011735 | 0.010820 | -0.001830 | 2.7816E-05 |
| H38 | 0.024800 | 0.029324 | 0.033848 | 0.009048 | -0.00013753 |
| H40 | 0.020455 | 0.022017 | 0.023579 | 0.003124 | -4.7485E-05 |
| H43 | 0.015812 | 0.017665 | 0.019518 | 0.003706 | -5.6331E-05 |
| H44 | 0.005652 | 0.006908 | 0.008163 | 0.002511 | -3.8167E-05 |
| H45 | 0.004232 | 0.005996 | 0.007760 | 0.003528 | -5.3626E-05 |
| N27 | 0.007171 | 0.004416 | 0.001661 | -0.005510 | 8.3752E-05 |
| O11 | 0.069724 | 0.066168 | 0.062613 | -0.007111 | 0.00010809 |
| O13 | 0.029320 | 0.025738 | 0.022155 | -0.007165 | 0.00010891 |
| O15 | 0.037285 | 0.051372 | 0.065458 | 0.028173 | -0.00042823 |
| O16 | 0.039352 | 0.032372 | 0.025391 | -0.013961 | 0.00021221 |
| O41 | 0.018685 | 0.020916 | 0.023146 | 0.004461 | -6.7807E-05 |
| O7 | 0.054304 | 0.036814 | 0.019324 | -0.034980 | 0.0005317 |
Adicionalmente, los descriptores duales confirman regiones de ambivalencia química en sitios como C17 y C2, donde los valores de 𝑓+ y 𝑓− son muy similares de 0.00001 a.u. y 0.00002 a.u., sugiriendo su capacidad para actuar como nucleófilos o electrófilos dependiendo del contexto de la reacción. Finalmente, estos descriptores locales complementan la caracterización global del sistema molecular al proporcionar información detallada de los puntos más reactivos y sus posibles interacciones, permitiendo una predicción precisa de las rutas de reacción más probables.
Estudios de anclaje molecular
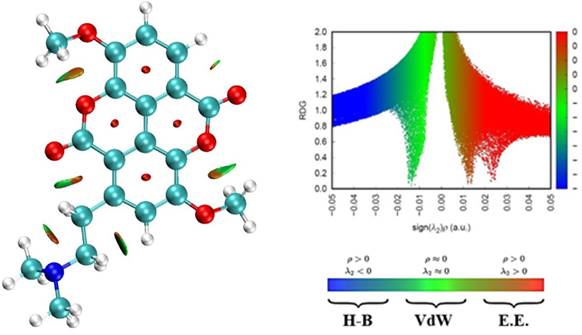
La búsqueda de las posibles Dianas mediante el SwissTargetPrediction17, dio como resultado un listado de enzimas candidatas, dentro de las cuales se encuentran los receptores acoplados a proteínas G, kinasas, proteasas e hidrolasas. El programa elige en función de la similitud molecular y la alta probabilidad estadística dada por la herramienta bioinformática que seleccionó a la acetilcolinesterasa (AChE-S) clasificada como hidrolasa (figura 2).
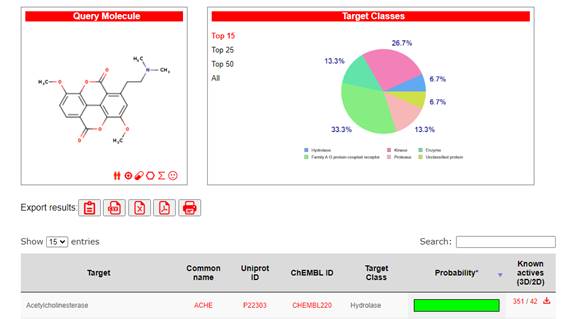
Figura 2 Interfaz del servidor SwissTargetPrediction donde se muestra a la taspina con los diferentes tipos de Dianas seleccionadas.
El análisis de la estructura de la AChE empleando el servidor en línea CASTP18, evidenció una cavidad cuyo sitio activo presentó un volumen de 933 Å3 y una superficie de 599.6 Å2 (Figura 3). En esta región con el programa UCSF Chimera19, se identificó a los aminoácidos terminales activos que aparecen de color verde en la figura. Estos aminoácidos fueron ASN 233, PRO 235, GLU 313, ILE 316, ASN 317, VAL 367, VAL 370, HIS 405, CYS 409, PRO 410, ALA 412, GLN 413, GLY 416, ARG 417, TYR 503, ALA 505, GLN 508, LEU 524, ALA 526, CYS 529, ALA 530, TRP 532, ASN 533, ARG 534, LEU 536, PRO 537, LYS 538 y LEU 540.
Figura 3 (a) Cavidad seleccionada por el CASTP de la AChE de color rojo. Figura 3 (b) Secuencia completa de aminoácidos de la AChE-S, donde los de color verde corresponden a los aminoacidos terminales activos.
Respecto a las simulaciones por anclaje molecular que se realizaron 50 veces entre la taspina y la enzima acetilcolinesterasa presentaron en promedio una energía mínima de unión -7.1 Kcal/mol, y al realizar el anclaje molecular flexible la energía llegó hasta los -8.71 Kcal/mol, lo cual significa que la formación del complejo taspina/acetilcolinesterasa es estable, favorable y espontánea. El análisis post docking se realizó con el Discovery Studio Visualizer V24 [20], con el cual se identificaron interacciones más importantes en el sitio activos correspondientes a los aminoácidos PRO 290, ARG 296, VAL 239, GLY 240, PRO 235 y HIS 405 (figura 4). Así como la estructura de la superficie hidrofóbica (figura 5) donde se aprecia la cavidad en la cual se fijó la taspina durante el acoplamiento molecular.
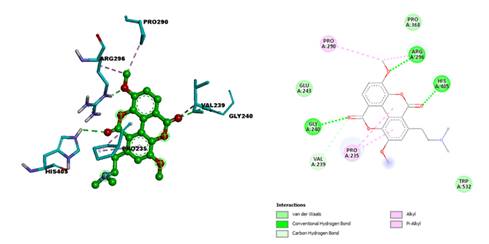
Figura 4 En la imagen de la izquierda se muestran las interacciones de los aminoácidos terminales más representativos con la taspina. En la imagen de la derecha aparecen los diferentes tipos de interacciones no covalentes más importantes en el momento del acoplamiento.
En el programa también se identificaron interacciones no covalentes que influyeron en el valor de la energía mínima de unión en el complejo (Figura 4). Las más importantes fueron el convencional puente de hidrógeno, enlace de carbono-hidrógeno, alquil y pi-alquil. En la tabla 2, se registraron junto con las distancias a la cual actuaron frente al ligando.
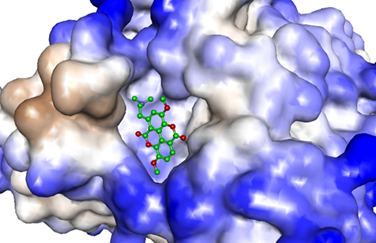
Tabla 3 Listado de los aminoácidos terminales que interactuaron con la AChE.
| Categoría | Inicio | Final | Distancia (A°) | |
| Enlace de hidrógeno | GLY 240: H (dador de enlace hidrógeno) | Taspina: O (Aceptor de enlace) | 2.162 | |
| AChE/Taspina | Enlace de hidrógeno | ARG 296: H (dador de enlace hidrógeno) | Taspina: O (Aceptor de enlace) | 2.131 |
| Enlace de hidrógeno | HIS 405: H (dador de enlace hidrógeno) | Taspina: O (Aceptor de enlace) | 2.910 | |
| Enlace de hidrógeno | VAL 239: C (dador de enlace hidrógeno) | Taspina: O (Aceptor de enlace) | 3.324 | |
| Hidrofóbico | Taspina: C (Alquil) | PRO 290 (alquil) | 4.594 | |
| Hidrofóbico | Taspina: C (alquil) | ARG 296 (alquil) | 4.161 | |
| Hidrofóbico | Taspina (orbitales-pi) | PRO 235 (alquil) | 5.238 | |
| Hidrofóbico | Taspina (orbitales-pi) | PRO 235 (alquil) | 5.441 |
Estudios de simulación de Dinámica Molecular (DM)
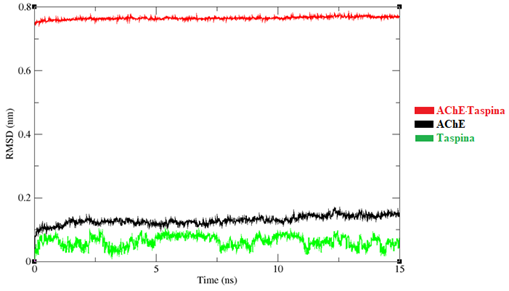
Se utilizó la mejor posición del análisis de docking molecular y se realizó la simulación de DM all-atoms en un periodo de 15ns a temperatura fisiológica. Las interacciones de la AChE y taspina se analizaron mediante la investigación de los gráficos de RMSD, Rg, SASA y RMSF, como se muestran en las figuras 6-9. La Figura 6, muestra que el RMSD del complejo AChE-Taspina presenta un equilibrio estable a aproximadamente 0.7 - 0.9 nm, estos valores pueden deberse a factores como flexibilidad intrínseca de la proteína, o cambios conformacionales en esta. El backbone de la proteína con RMSD en 0.2 - 0.3 nm indica que la estructura es bastante estable a lo largo de la simulación, lo que nos indica que la proteína no ha sufrido grandes cambios conformacionales, al menos en su cadena principal. El ligando con un RMSD 0.1 - 0.2 nm indica que ha tenido una desviación mínima durante la simulación, lo que sugiere que el ligando ha mantenido una posición fija con respecto a la posición inicial, mostrando una fuerte interacción entre el ligando y la proteína.
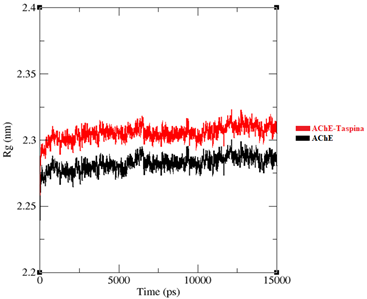
En la figura 7 , se determinó el radio de giro del sistema a lo largo de 15 ns, donde nos muestra que ambas curvas estabilizan desde el inicio y no exhiben grandes fluctuaciones, lo que nos indica que la proteína y el complejo han alcanzado un equilibrio sin presentar grandes cambios conformacionales, demostrando flexibilidad dinámica de la proteína y su interacción con el ligando. En la proteína, el radio de giro se encuentra entre 2.25 - 2.3 nm, lo que indica una estructura bastante compacta. Con respecto al complejo, se observa que presenta un valor ligeramente más alto, con un valor de 2.3 - 2.35 nm, lo que sugiere que el ligando aumenta el tamaño del sistema, pero sin una contribución significativa por un buen acoplamiento a la proteína.
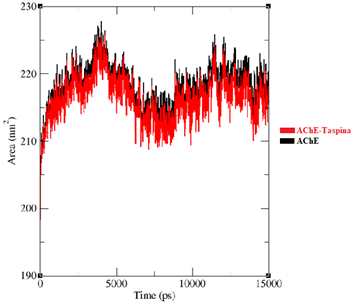
La figura 8, nos muestra el análisis del Área Superficial Accesible al Solvente (SASA) que mide el área de la superficie de la molécula que está expuesta al solvente16. Las fluctuaciones que se observan son típicas en una simulación de dinámica molecular que corroboran que no hubo cambios drásticos en la exposición al solvente a lo largo del tiempo de simulación. Este gráfico denota que después de los 5ns tanto el complejo como el backbone alcanzan un equilibrio, con fluctuaciones menores, lo que indica que la proteína y el ligando han alcanzado un estado estable en términos de interacción con el solvente. Adicionalmente, observa que el SASA del complejo es un poco menor al backbone, esto sugiere que el ligando está bien acoplado a la proteína y posiblemente protegido por la estructura proteica, esto es un indicio favorable entre el ligando y proteína, denotando que el ligando no está demasiado expuesto al solvente.
En la figura 9, se representa el análisis RMSF que nos permite evaluar la flexibilidad de cada residuo de la proteína a lo largo de la simulación. Ambas curvas muestran valores de RMSF bajos, lo que indica que la proteína ha permanecido bastante estable durante la simulación, denotando ser un buen indicador de estabilidad en la dinámica. Se observan dos picos (el primero y último) con elevados valores de RMSF que alcanzan los 0.5 - 0.6 nm. Siendo esto común en las proteínas, ya que los extremos suelen ser más flexibles y no están restringidos estructuralmente. Aunque las dos curvas están superpuestas, los valores de curva roja tienden a ser ligeramente más altos, lo que indica que la presencia del ligando puede estar contribuyendo a fluctuaciones adicionales en la estructura de la proteína. Esto una vez más nos muestra que la simulación de dinámica molecular ha sido bien equilibrada y que la proteína ha mantenido su estructura durante la simulación.
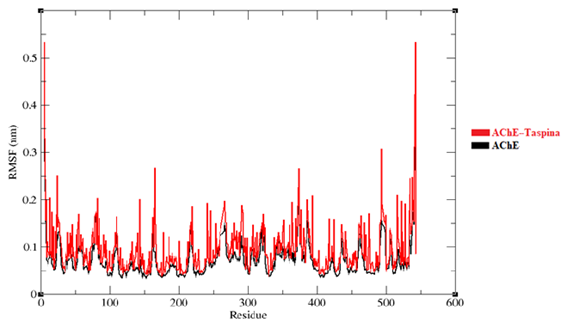
Estos análisis de la simulación de Dinámica Molecular revelan en conjunto una estabilidad estructural entre el AchE y taspina, lo que sugiere que podría ser un buen inhibidor potencial de la enzima, ya que podría estar bien acoplada al sitio de unión.
CONCLUSIONES
El análisis mediante el método de gradiente reducido de la densidad (RDG) confirma la ausencia de enlaces por puente de hidrógeno, pero resalta la presencia de interacciones Van der Waals en la región ecuatorial y repulsiones estéricas en los anillos aromáticos. Los descriptores globales indican una moderada estabilidad electrónica y una tendencia a comportarse como nucleófilo. A nivel local, el análisis de la función de Fukui revela que algunos carbonos y oxígenos específicos son los sitios más reactivos para ataques nucleofílicos y electrófilos.
Los resultados obtenidos de las simulaciones de docking molecular entre la taspina y la enzima acetilcolinesterasa, con una energía mínima de unión de -8.71 kcal/mol, confirman la estabilidad y espontaneidad del complejo. Estos valores indican que la interacción es termodinámicamente favorable, lo que sugiere un buen ajuste entre la taspina y el sitio activo de la enzima. Los residuos clave responsables de las interacciones más significativas son PRO 290, ARG296, VAL239, GLY240, PRO235 y HIS 405, proporcionan información crítica para entender los mecanismos moleculares de inhibición de la acetilcolinesterasa por la taspina. Además, la cavidad identificada en la superficie hidrofóbica de la enzima refuerza el acoplamiento estable y específico de la taspina, lo cual abre nuevas oportunidades para el diseño de inhibidores basados en este modelo de interacción.
El estudio de dinámica molecular revela que la interacción entre la AChE y taspina presenta un acoplamiento favorable y estructura proteica estable durante el tiempo de simulación. Esto fue corroborado con análisis de RMSD, Rg, SASA y RMSF, que muestran a la taspina como un inhibidor potencial de la enzima AChE debido a la limitada exposición al solvente de esta y su interacción estable, lo que sugiere una buena afinidad al sitio de unión.

