










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista Peruana de Biología
On-line version ISSN 1727-9933
Rev. peru biol. vol.15 no.2 Lima 2008
TRABAJOS ORIGINALES
Identificación in silico de un grupo de secuencias ortólogas conservadas (COS) de Ipomoea batatas
In silico prediction of conserved ortholog set (COS) sequences from Ipomoea batatas
Christian Solís-Calero
Centro Internacional de la Papa (CIP), P.O. Box 1558, Lima 12, Perú. Email Christian Solis Calero: csolis@esan.org.pe
Resumen
En el presente trabajo se describe una serie de procedimientos bioinformáticos para la predicción de un grupo secuencias ortólogas conservadas (COS) de Ipomoea batatas, así como la evaluación de su potencial utilidad para la generación de marcadores moleculares y estudios de diversidad en esta especie. Con ese propósito usando los programas BLAST X y TBLASTN se realizó una comparación reciproca por similaridad entre secuencias ESTs procedentes de librerías de cDNAs de Ipomoea batatas, propias o disponibles de modo público en la base de datos GenBank, con secuencias COS de Arabidopsis thaliana. La anotación funcional de las secuencias COS predichas en Ipomoea batatas se realizo usando los programas BLASTX, INTERPROSCAN y PSI-BLAST. Se obtuvieron en total 204 secuencias COS candidatos de Ipomoea batatas, siendo 16 secuencias provenientes de una librería generada a partir de raíces de reserva. Se evaluó de modo computacional el polimorfismo de las secuencias COS de raíces de reserva, obteniéndose SNPs en 8 secuencias, y secuencias repetidas en tandem en una de ellas.
Palabras clave: COS, Ipomoea batatas, genes de baja copia, ortólogos, ESTs.
Abstract
We develop some bioinformatics procedures to predict Conserved Ortholog Set (COS) sequences from Ipomoea batatas, and evaluate their usefulness for molecular markers and diversity studies of this species. We predict orthology relationship between Ipomoea batatas ESTs sequences and Arabidopsis thaliana COS sequences, according to Best Bidirectional Hits (BBHs) criteria, realizing similarity comparison using BLAST X and TBLASTN programs. We obtained a set of 204 putative COS sequences, 16 of them belonged to storage roots. Functional annotation of sweet potato predicted sequences COS was realized using BLASTX, INTERPROSCAN and PSI-BLAST programs. We evaluate possible polymorphisms in COS candidate sequences, finding SNPs in eight sequences and tandem repeats in one of them.
Keywords: COS, Ipomoea batatas, ortholog, low copy genes, ESTs.
Introducción
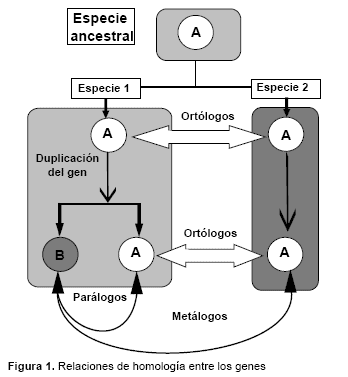
Los ortólogos son definidos como genes de diferentes especies que comparten un ancestro común por especiación. Por contraste, los genes parálogos son copias duplicados dentro de un genoma que pueden ser producidos por el fenómeno de poliploidización o duplicaciones en tandem (Gogarten & Olendzenski, 1999, Sonnhammer & Koonin, 2002) (Fig. 1).
Actualmente, debido a la disponibilidad cada vez mayor de bases de datos de secuencias genómicas y de ESTs, la predicción de la relación de ortología entre genes de diferentes especies puede ser realizada mediante métodos bioinformáticos, incluso entre genes de especies tan divergentes en las que por estudios experimentales antes no se podía predecir tal relación (Pennisi, 1998, Ku et al., 2000). Basados en ello Fulton et al. (2002) introdujeron el concepto de grupo de ortólogos conservados (COS), que son secuencias marcadoras obtenidas del procesamiento de la información de las bases de datos de secuencias biológicas. Estas secuencias corresponden a genes de plantas que se han mantenido conservados y en un bajo número de copias en sus genomas, desde los últimos ancestros comunes que comparten las plantas con flores. El diseño de iniciadores de PCR sobre estas secuencias puede permitir su uso para la obtención de marcadores moleculares basados en secuencias Ej: (CAPs, SSRs) y estudios de diversidad, incluso en especies relacionadas a la de origen de la secuencia (Kozik & Michelmore, 2002).
Los esfuerzos para la generación de nuevos marcadores moleculares en Ipomoea batatas son importantes, porque permitirían aumentar la densidad de los mapas genéticos recién generados en esta especie. Las secuencias COS a su vez pueden ser la base para estudios de evolución molecular que en Ipomoea batatas puedan determinar de modo más preciso su biogeografía y sus centros de origen. Otro de los propósitos de este trabajo es presentar un ejemplo de como se puede aprovechar la gran cantidad de información de secuencias depositada en las bases de datos biológicas, así como programas bioinformáticos, que son accesibles libremente por toda la comunidad científica a través de Internet, o pueden ser escritos según los requerimientos específicos de la investigación usando lenguajes de programación como Perl y PHP (Stajich et al., 2002).
Materiales y métodos
Bases de datos de ESTs de Ipomoea batatas
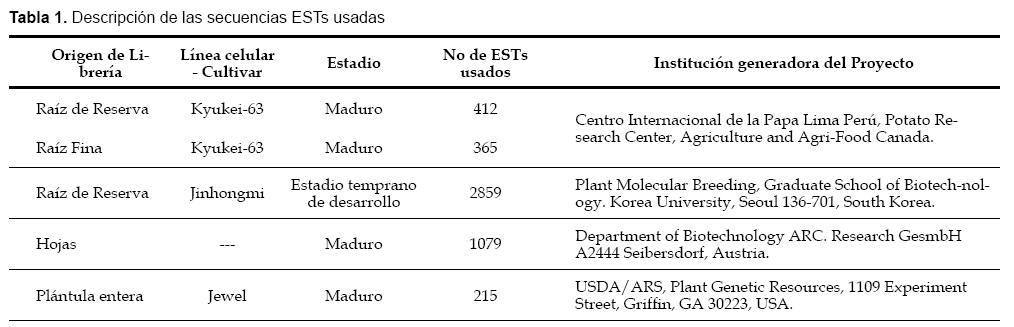
Se obtuvieron secuencias ESTs correspondientes a varias librerías de ESTs de Ipomoea batatas a través del National Center for Biotechnology Information (NCBI: http://www.ncbi.nlm.nih.gov ).
Asimismo se dispuso de secuencias ESTs pertenecientes a 2 librerías generadas en proyectos del CIP. La información sobre las librerías usadas se encuentra descrita en la Tabla 1. Las secuencias COS de Arabidopsis thaliana fueron obtenidas del Compositae Genome Project Database (Kozik & Michelmore, 2002). Para disminuir la redundancia de secuencias semejantes en cada grupo de ESTs, estas secuencias según su origen fueron sometidas a un proceso de ensamblaje usando el programa CAP3 (Huang & Madan, 1999), obteniéndose como resultado contigs en el caso de secuencias redundantes, y singletons si las secuencias eran únicas. Las secuencias ESTs, así como la información derivada de su procesamiento fueron depositadas en una base de datos llamada Kumara ESTs, desarrollada usando el paquete de programas XAMPP (que incluye Apache, MySQL, PHP y Perl).
Determinación de secuencias ortólogas
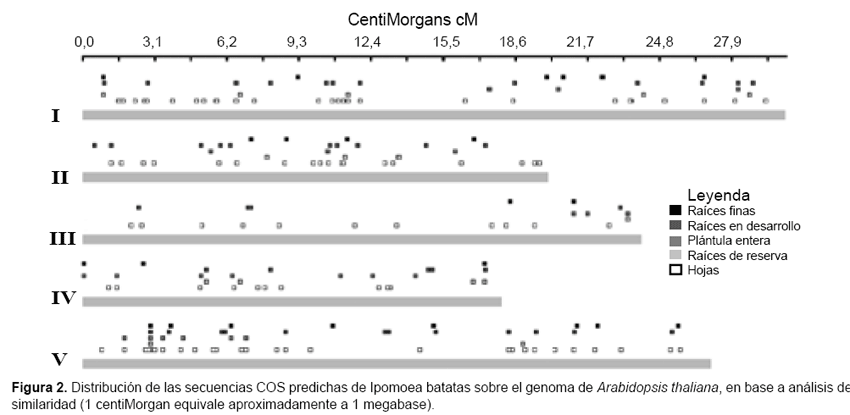
Para la identificación de ortólogos se siguió el criterio de Best Bidireccional Hits (BBHs) (8); se realizaron dos búsquedas de similaridad entre las secuencias ESTs de Ipomoea batatas y las secuencias de aminoácidos correspondientes a los genes COS de Arabidopsis thaliana, usando localmente los programas BLAST-X y TBLASTN. Los parámetros de búsqueda fueron en ambos casos W= 12, matriz: BLOSUM62, T(thereshold)= 11, mínimo valor de E= 10-6, tamaño teórico del query= 1000. Los resultados de la búsqueda BLAST fueron posteriormente resumidos usando un programa en Perl escrito por nosotros. Se seleccionaron como secuencias COS candidatos, las secuencias ESTs que presentaron en los alineamientos con las secuencias COS de Arabidopsis, valores E inferiores a 10-6 y porcentajes de identidad superiores al 30% y longitudes de alineamiento mayor igual a 70 aminoácidos y que no presentaron similaridad significativa con más de una secuencia de Arabidopsis. Los resultados de los análisis de similaridad fueron procesados mediante otros programas Perl previamente escritos, obteniendo los archivos de entrada del programa Genome Pixelizer (Kozik et al., 2002), que permitió visualizar la distribución de las secuencias COS candidato de Ipomoea batatas sobre el genoma de Arabidopsis thaliana.
Anotación Funcional
La anotación funcional de las secuencias COS predichas en Ipomoea batatas se realizó usando localmente el programa BLASTX (Altschul et al., 1997), y de modo online mediante los programas INTERPROSCAN (Quevillon et al., 2005) localizado en: http://www.ebi.ac.uk/InterProScan/ , y PSI-BLAST (Altschul & Koonin, 1998) localizado en: http://www.ncbi.nlm.nih.gov/BLAST/ .
Predicción de polimorfismos en las secuencias COS candidato
Con el objetivo de evaluar la posible utilidad de las secuencias COS obtenidas para la generación de marcadores moleculares y estudios de diversidad, se realizó la predicción computacional de polimorfismos de tipo SNPs y repeticiones en tandem en el interior de las secuencias COS correspondientes a las secuencias contigs de raíces de reserva.
Cada secuencia se comparó contra la base de datos EST del NCBI usando el programa BLASTN. De esta base de datos Se obtuvieron las secuencias de Ipomoea batatas cuyos alineamientos presentaron valores de E menores a 10-30, y longitudes de alineamiento que superaran el 80% de la longitud de la secuencia. Estas secuencias fueron agrupadas con los ESTs que formaban cada contig, y ensambladas usando el programa CAP3 (Huang & Madan, 1999). Usando los resultados del ensamblaje de secuencias, se realizo la predicción de: SNPs usando el programa AutoSNP (Barker et al., 2003); microsatélites con el programa Sputnick (Jewell et.al, 2006); y de las repeticiones en tandem con el programa Tandem Repeats Finder (Benson, 1999).
Resultados
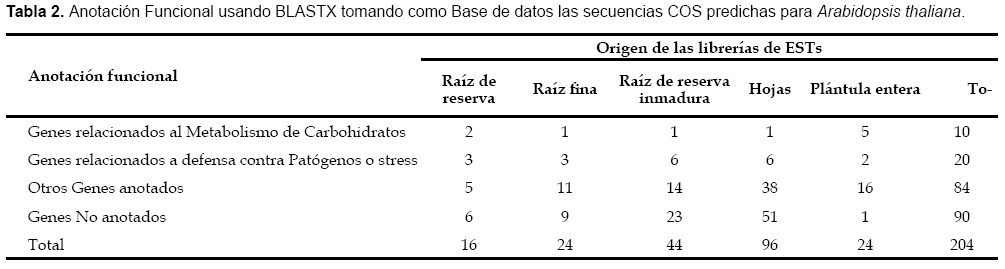
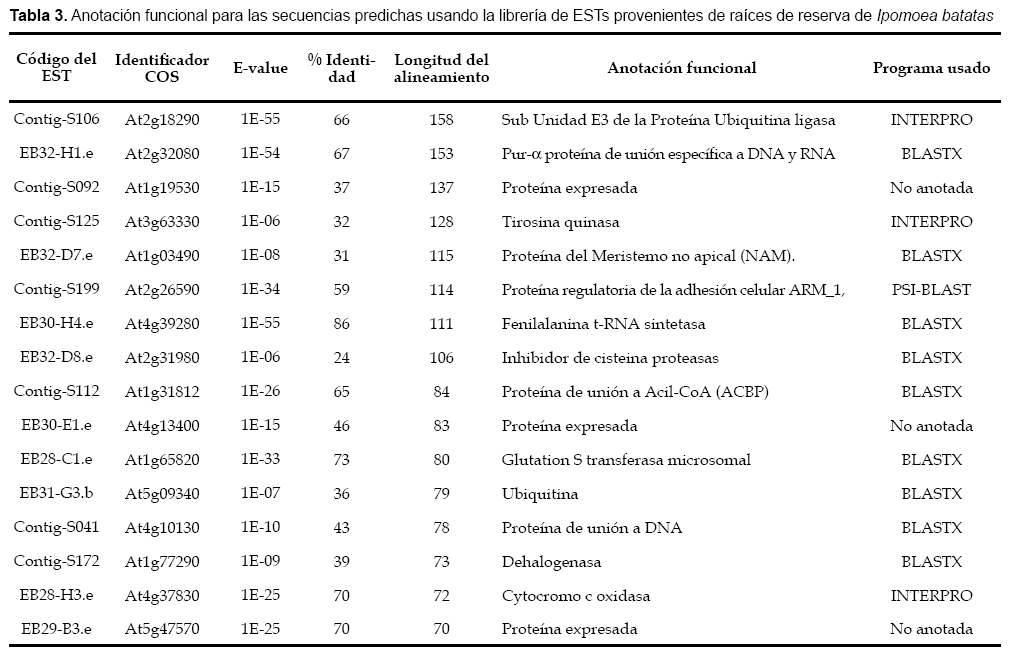
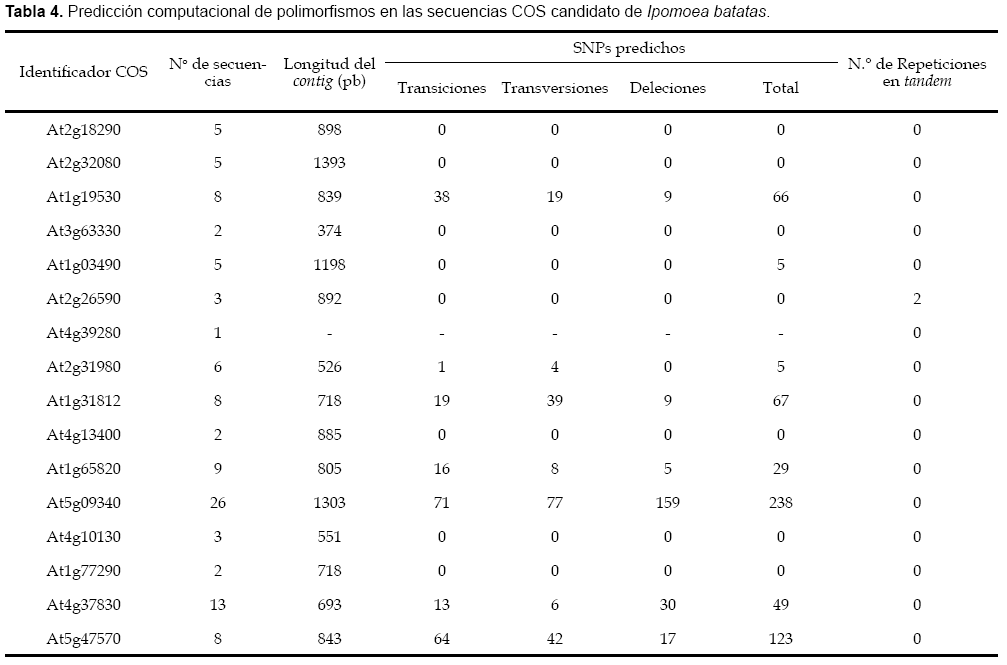
Se obtuvieron en total 204 secuencias COS candidatos de Ipo- moea batatas, siendo 16 secuencias provenientes de la librería generada a partir de raíces de reserva, los detalles se muestran en las tabla 2 y tabla 3. La distribución de todas las secuencias COS candidato de Ipomoea batatas sobre el genoma de Arabidopsis thaliana se pueden visualizar en la figura 2. Solo se pudo predecir polimorfi smos de secuencia SNPs en ocho secuencias, y de secuencias repetidas en tandem en solo una secuencia, los detalles se muestran en la tabla 4.
Discusión
La defi nición de secuencias COS considera que sean genes de baja copia, y es por esa característica que estas secuencias pueden ser de utilidad en estudios de evolución molecular, biodiversidad y generación de marcadores moleculares (Fulton et.al., 2002).
Para asegurar esta característica, en la predicción de secuencias COS propias de camote hemos usado como referencia las secuencias COS de Arabidopsis thaliana, en la que por análisis de su genoma completo, se ha determinado que esas secuencias son de baja copia (Kozik & Michelmore, 2002). La relación de ortología se estableció entre las secuencias COS de Arabidopsis y secuencias ESTs de Ipomoea batatas usando el criterio de Best Bidireccional Hits (BBHs. Según esta definición, dos genes, de dos organismos diferentes, respectivamente, son ortólogos si luego de una búsqueda de similaridad en ambos genomas ambas muestran ser en forma reciproca, más similares entre si respecto a otros genes (Zheng et al., 2005). A esta definición nosotros incluimos tres restricciones más: que la similaridad tenga valores de E-value superiores a 1x10-6, un porcentaje de identidad superior al 25% y una longitud de alineamiento mayor o igual a 70 aminoácidos. Hemos considerado estos valores porque son cercanos a los sugeridos por otros autores (Claverie & Notredame, 2003), porque la longitud mínima aceptable para un EST es de 200 pb (cercano al valor de 70 aminoácidos), y porque disminuyen la probabilidad de registrar como COS a secuencias que presentan similaridad pero solo por compartir dominios comunes a varias proteínas y que no necesariamente revelan que las secuencias comparadas tengan una relación de homología (ancestro común).
Debemos considerar que los linajes de Arabidopsis thaliana e Ipomoea batatas se separaron hace 100 a 150 millones de años (14) por lo que la relación de similaridad entre dos genes ortólogos no tiene que ser necesariamente alta. Por ello hemos empleado una comparación DNA (ESTs) - proteínas (Secuencia COS Arabidopsis). Ésta comparación a diferencia de las comparaciones DNA-DNA, conservan mas información y puede predecir homologías más remotas.
En la tabla 2 podemos observar que cerca al 50% de las secuencias COS predichas en Ipomoea batatas (90 de 204) no presentan una anotación funcional luego de la búsqueda de similaridad usando Blast X, y es porque estos métodos de comparación de pares de secuencias presentan serias limitaciones para encontrar homólogos remotos, que son aquellos que comparten un mismo origen evolutivo pero que han divergido mucho y su identidad de secuencia está por debajo del 25% (Mount, 2003). Por ello para completar la anotación funcional hemos empleado un metaservidor como el INTERPROSCAN, el cuál integra en una sola búsqueda a muchas bases de datos de patrones, perfiles, dominios y motivos que permiten realizar la búsqueda de similaridad, pero restringida a residuos funcionales de la proteína (centros activos, zonas reguladoras), que han sido conservados durante la evolución por su importancia (Quevillon et al., 2005).
Otro método usado para la anotación funcional fue PSIBLAST, programa que primero realiza una búsqueda BLAST de similaridad de secuencia en una base de datos, y a partir de los resultados, construye un perfil o PSSM (position specific scoring matrix). Posteriormente, usando este perfil hace una nueva búsqueda en la base de datos, encontrando idealmente nuevas secuencias homólogas remotas, cuya información permite generar un nuevo perfil, que a su vez permite realizar otra búsqueda para predecir nuevas secuencias homologas remotas (Altschul & Koonin, 1998).
En la figura 2 se observa que las 204 secuencias COS de Ipomoea batatas predichas presentan una distribución que cubre la mayor parte del genoma de Arabidopsis, observándose que en muy pocos casos los ESTs de las diferentes librerías de cDNA coinciden en algún punto del genoma, lo que se explica porque los ESTs provienen de tejidos bastante diferenciados no solo por su función sino incluso por su estadio de desarrollo. Es posible inferir por ello que los ESTs que cubren una misma posición deben corresponder a genes constitutivos de la planta.
La predicción computacional de polimorfismos en las secuencias COS candidato se hace a fin de seleccionar las secuencias en las que con mayor probabilidad hay variaciones que puedan expresarse en la obtención de marcadores moleculares a partir de un estudio que involucre el uso de DNA genómico de diferentes variedades o poblaciones de Ipomoea batatas. Esta metodología puede ser utilizada en diferentes organismos, pero depende de la disponibilidad de secuencias en las bases de datos biológicas para el análisis. No obstante, con el incremento de los proyectos de secuenciación de genomas y el continuo y abundante depósito de información de secuencia (Paterson et al., 2000), esta posibilidad se incrementa, por lo que los métodos presentados se pueden constituir sin problemas en un proceso rutinario de trabajo en los laboratorios de Genética Molecular.
Nuestros resultados (tabla 4) muestran que a mayor número de secuencias que forman un contig, se obtiene mayor número de polimorfismos de tipo SNPs, obteniéndose en la mayoría de casos un mayor número de polimorfismos por transiciones que transversiones y deleciones respectivamente, lo que esta de acuerdo con la tendencia esperada para las probabilidades de estos tipos de mutación, y son estas secuencias las que podrían ser seleccionadas para el diseño de iniciadores de PCR, que conduzcan a los experimentos para la obtención de marcadores moleculares.
En los casos en los que las deleciones superan a los otros tipos de variación, probablemente se puede deber a que las secuencias del GenBank incorporadas al análisis de predicción de polimorfismos no provienen de genes ortólogos, sino paralógos con una similaridad de secuencia bastante alta, y que no pudieron filtrarse previamente con los análisis de similaridad, lo que puede ser reflejo de un origen evolutivo reciente. Estas últimas secuencias serían muy difíciles de utilizar para la obtención de marcadores moleculares, porque probablemente los iniciadores de PCR diseñados en ellas podrían coincidir en varias regiones del genoma, amplificando varios genes a la vez y con ello haciendo difícil la interpretación de los resultados experimentales.
Literatura citada
Altschul S.F.&, E.V. Koonin. 1998. Iterated profile searches with PSI-BLAST--a tool for discovery in protein databases. Trends Biochem Sci.23(11):444-447.
Altschul S.F., T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller, & D.J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res; 25(17):3389-402.
Barker G, J. Batley, H. OSullivan, K.J. Edwards, D. Edwards. 2003. Redundancy based detection of sequence polymorphisms in expressed sequence tag data using autoSNP. Bioinformatics. 19(3):421-422.
Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27(2): 573-580.
Claverie J-M. & C. Notredame. 2003. Similarity searches on Sequence Databases Bioinformatics for Dummies, New York, Wiley Publishing Inc., pp 279-215
Fulton T.M., R. Van der Hoeven, N.T. Eannetta, & S.D. Tanksley. 2002. Identification, analysis, and utilization of conserved ortholog set markers for comparative genomics in higher plants. Plant Cell 14: 1457-1467.
Gogarten J.P., & L. Olendzenski. 1999. Orthologs, paralogs and genome comparisons. Curr Opin Genet Dev. Dec;9(6):630-636.
Huang, X. & A. Madan,. 1999 CAP3: a DNA sequence assembly program Genome Res; 9, 868–877
Jewell E, A. Robinson, D. Savage, T. Erwin, C.G. Love, G.A. Lim, X. Li, J. Batley, G.C. Spangenberg & D. Edwards. 2006. SSRPrimer and SSR Taxonomy Tree: Biome SSR discovery. Nucleic Acids Res. 34:W656-659.
Kozik A & R. Michelmore. 2002. Computational approach to select the set of ESTs with a single BLAST hit to Arabidopsis genome. University of California at Davis http://cgpdb.ucdavis.edu/COS_Arabidopsis/ (acceso 20/02/07)
Kozik A, Kochetkova E, Michelmore R. 2002. GenomePixelizer--a visualization program for comparative genomics within and between species. Bioinformatics. 8(2):335-336.
Ku H.M., T. Vision, J. Liu, & S.D. Tanksley. 2000. Comparing sequenced segments of the tomato and Arabidopsis genomes: large-scale duplication followed by selective gene loss creates a network of synteny. Proc Natl Acad Sci USA; 97(16):9121-91216.
Mount D. 2003. Bioinformatics: Sequence and Genome Analysis. Cold Spring Harbour Laboratory Press, 10 Skyline Drive, Plainview, New York 11803-2500.
Paterson A.H., J.E. Bowers, M.D. Burow, X. Draye, C.G. Elsik, C.X. & et al.. 2000. Comparative genomics of plant chromosomes. Plant Cell; 12(9):1523-1540.
Pennisi E. 1998. A bonanza for plant genomics. Science; 282(5389):652-4.
Quevillon E, V. Silventoinen, S. Pillai, N. Harte, N. Mulder, R. Apweiler, & R. Lopez. 2005. InterProScan: protein domains identifier. Nucleic Acids Res. Jul 1(33): 116-120
Sonnhammer E.L. & E.V. Koonin. 2002. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet;18(12):619-620.
Stajich J.E., D. Block, K. Boulez, S.E. Brenner, S.A. Chervitz, C. Dagdigian & et al. 2002. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 12(10):1611-1618.
Zheng X.H., F. Lu, Z.Y. Wang, F. Zhong, J. Hoover, & R. Mural. 2005. Using shared genomic synteny and shared protein functions to enhance the identification of orthologous gene pairs. Bioinformatics; 21(6):703-710
_________________
Presentado: 22/02/2007
Aceptado: 06/01/2008
