











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La enfermedad inflamatoria intestinal (EII) se caracteriza por ser un proceso inflamatorio, crónico, recurrente, con diferentes grados de severidad que afecta el tubo digestivo, pero que además puede afectar otros órganos. Dentro de la EII se clasifican la enfermedad de Crohn (EC) y la colitis ulcerosa (CU) 1. Esta patología suele tener dos picos de presentación, uno en el segundo, y otro en la tercera década de la vida 2. Esta inflamación crónica es caracterizada por ser de tipo netamente mucoso, simétrico y solo comprometiendo el intestino grueso en la CU y transmural, focal, asimétricos y con posibilidad de comprometer cualquier trayecto del tubo digestivo en la EC 1. Las manifestaciones clínicas son heterogéneas, dependiendo del segmento comprometido, usualmente el paciente se queja de dolor abdominal, diarrea crónica, algunas veces con rectorragia, urgencia, tenesmo, pérdida de peso, anemia, sin olvidar que también puede presentar compromiso extraintestinal dentro del cual los más frecuentes son el articular, cutáneo y ocular. Dentro de las complicaciones está el compromiso fistulizante, perianal y estenosante principalmente presentado en la EC 3.
Existen aproximadamente 1,4 millones de personas afectadas con EII en Estados Unidos y Canadá, y 2,2 millones en Europa y su morbimortalidad es grande 4,5. En Colombia ya existen 2 estudios de prevalencia de EII el primero en el 2017 documento CU del 51,77 / 100 000 y de EC de 5,85 / 100 000 6, en el 2020 se incrementó en CU a 113,12 / 100 000 y en EC a 16,57 / 100 000 7.
Se han hecho algunos estudios para tratar de identificar los factores genéticos que confieren susceptibilidad para EII. Hasta la fecha, se han documentado más de 240 loci 8 únicos asociados a EII en más de 50 genes, gracias a estudios de asociación de todo el genoma (Genome Wide Association Studies, GWAS por sus siglas en inglés), principalmente en población adulta 9,10. La mayoría de estos loci de susceptibilidad identificados parecen tener individualmente un efecto bajo (OR ~ 1,0-1,5), y en conjunto parece que representan menos del 20% del riesgo hereditario para EII 11. Lo anterior apoya un modelo de patología complejo, en el que variantes comunes con efectos modestos parecen interactuar con factores ambientales como la dieta, el tabaquismo y el microbioma intestinal, dando lugar a la susceptibilidad a EII. Por lo tanto, se acepta que la EII no sigue un patrón de herencia mendeliano sino que es el resultado de una respuesta inflamatoria inapropiada del microbioma intestinal en individuos genéticamente susceptibles 12.
Algo que apoya la heredabilidad y el componente genético de esta patología, es que se ha documentado que hasta un 20% de individuos tienen historia familiar de EII. Y que además, en el 25-35% de estas familias, existen individuos afectados con ambas entidades (EC y CU). Parientes en primer grado de pacientes con CU o EC tienen un riesgo hasta 10 veces mayor de presentar EII 13. En EC, la concordancia en gemelos monocigóticos es del 30.3%, y en dicigóticos del 3,6%. En CU, la concordancia en gemelos monocigóticos es del 15,4% y en dicigóticos del 3,9%. Los estudios en gemelos han demostrado la mejor evidencia de la predisposición genética a EII, que será mayor para EC que para CU 14.
Así mismo, se han documentado también síndromes genéticos asociados a EII, como el síndrome de Turner, el síndrome de Hermansky -Pudlak, enfermedad de células falciformes, trisomía 9, deficiencia selectiva de IgA, síndrome de Chediak Higashi, y paquidermaperiostosis, entre otros 15.
Es importante resaltar que hay genes como el NOD2, MDR1, CARD15 que parecen afectar directamente la respuesta inmunológica normal en el intestino 16,17. Recordemos que, en estado saludable, las células caliciformes secretan una capa de moco que limita la exposición del epitelio intestinal a bacterias. Tanto la secreción de péptidos antimicrobianos (por ejemplo, Α-defensinas) por las células de Paneth como la producción de inmunoglobulina A (IgA) proporcionan protección adicional contra el microbioma luminal 18. La detección microbiana innata por las células epiteliales, las células dendríticas y los macrófagos está mediada por receptores de reconocimiento de patrones, como los receptores tipo toll y las proteínas del dominio de oligomerización de nucleótidos (NOD). Las células dendríticas presentan antígenos a las células T CD4 + vírgenes en órganos linfoides secundarios (placas de Peyer y ganglios linfáticos mesentéricos), donde factores como el fenotipo de las células presentadoras de antígeno y el medio de citocinas (factor de crecimiento transformante β [TGF-β] e interleucina - 10) modulan la diferenciación de subgrupos de células T CD4 + con la producción de perfiles característicos de citocinas (células T reguladoras por ejemplo, Treg, Th1, Th2 y Th17]) y moléculas enterotrópicas como por ejemplo, α4β7 que proporcionan la localización intestinal de los linfocitos de la circulación sistémica. Estas células T CD4 + activadas luego circulan hacia la lámina propia intestinal, donde llevan a cabo funciones efectoras 19. Teniendo en cuenta esta inmunología de mucosas, también se han documentado alteraciones en ciertos genes pueden involucrar cambios en este proceso, incluso se han implicado algunos genes en el pronóstico de la enfermedad como lo son:
• NOD2: se ha asociado a un incremento del riesgo para EC de tres veces más con respecto a la población general, si hay variantes heterocigotas, 38 veces más si variantes homocigotas y 44 veces más si variantes en heterocigosis compuesta 20,21. Está asociado a mayor agresividad en EC, mayor riesgo de enfermedad estenosante y necesidad de intervención quirúrgica temprana 22.
• ABCB1: algunos polimorfismos se han asociado a incremento en el riesgo para CU 23.
Hay que resaltar que también hay implicaciones terapéuticas en las alteraciones genéticas. Se han documentado alteraciones en los genes ATG16L1, NOD2 e IRGM, los cuales están involucrados en aumentar las vías de autofagia con posterior evidencia de no eficacia para algunos medicamentos. Por eso, gracias a la identificación de nuevos loci de riesgo, se han diseñado medicamentos dirigidos que permiten el acercamiento a la medicina personalizada 24.
Se requiere un cambio de perspectiva para avanzar en la comprensión de la EII. La mayoría de enfoques de investigación tradicionales se han centrado en aspectos de la etiología o tratamiento, sin una apreciación de las asociaciones etiológicas y farmacológicas complejas. Mediante el siguiente estudio se propone realizar una caracterización fenotípica y genotípica de pacientes con EII en población colombiana y describir su posible asociación con predisposición.
MATERIALES Y MÉTODOS
Diseño del estudio y pacientes
Estudio observacional descriptivo tipo serie de casos, el análisis de la frecuencia de estas variantes se hizo teniendo en cuenta las frecuencias alélicas reportadas en la literatura y los datos arrojados por el proyecto 1000 genomas.
Se incluyeron 16 pacientes seleccionados por conveniencia de la Consulta de Enfermedad Inflamatoria Intestinal del Hospital Universitario San Ignacio. Todos tuvieron asesoramiento genético pre test y se realizaron los árboles genealógicos de mínimo tres generaciones de sus familias con el objetivo de establecer antecedentes de patologías asociadas con sistema gastrointestinal o enfermedades autoinmunes. Se incluyeron pacientes diagnosticados con EC o CU por criterios clínicos y anatomopatológicos de acuerdo a biopsia intestinal, con inicio de síntomas gastrointestinales después de los 18 años.
Control de sesgos
Dado que se trata de un estudio observacional y retrospectivo, los datos fueron tomados de la historia clínica. El control de sesgo de información se realiza teniendo en cuenta que los pacientes pertenecen a la clínica de EII del Hospital Universitario San Ignacio. Por lo tanto, son pacientes con un seguimiento médico continuo, en quienes se realiza control paraclínico seriado y registro de signos y síntomas en varias consultas y evoluciones de la historia clínica, por lo tanto, la consolidación de las variables tiene alta fidelidad y representatividad disminuyendo sustancialmente el sesgo de memoria y de registro de variables.
Tamaño de muestra
Como es una serie de casos no se tiene un tamaño de muestra dado que no se quiere realizar una inferencia de los resultados teniendo en cuenta una variabilidad de condiciones clínicas que no permiten la generalización de los resultados de estos pacientes, pero sí describir asociaciones y características demográficas con los hallazgos genéticos y fenotípicos en estos pacientes. Como es un estudio en el que se requiere inversión económica para poder realizar el estudio de los paneles genéticos, hay una limitación en la inclusión de más pacientes, el cual estuvo condicionado al presupuesto disponible.
Extracción de ADN y genotipificación
Se extrajeron muestras de ADN de sangre total de cada paciente mediante el método de Salting out en el Laboratorio del Instituto de Genética Humana de la Pontificia Universidad Javeriana Bogotá. Todas las muestras de ADN se genotipificaron utilizando un panel de genes múltiples basado en secuenciación de próxima generación (NGS por sus siglas en inglés) que incluía 46 genes relacionados con EII y algunos trastornos relacionados con autoinmunidad: IL23R, IL12B, JAK2, TYK2, STAT1, STAT3, STAT4, IBD3, REL, CARD9, ITAM, IL2RA ICOSLG, PRDM1, SMAD3, ORMDL3, CD48, STAT1, IL21, DOK3, CREM, CD226, MLH3, IFNG, CD6, MAP3K8, SLC11A1, NOD2, IL6, IBD5, NKX2-3, TNF, CCR6, IBD5, IL1R2, IRGM, ATG16L1, PTPN22, IL6ST, SPRED1, IFNGR2, TNFSF18, TNFRSF14, CARD11 e IRF5.
Análisis genómico de las variantes
Se aplicaron varios filtros de calidad a las variantes que se encontraron, incluidas aquellas con bajas frecuencias o ausentes de los controles. Para el análisis descriptivo de los single nucleotide polymorphism (SNP) se utilizó la Nomenclatura Internacional del Genoma Humano (HUGO-GNC: Comité de Nomenclatura Genética de la Organización del Genoma Humano) y adicionalmente se les asignó significación clínica según las bases de datos OMIM, ClinVar y dbSNP. Se realizaron algoritmos in sílico: Polyphen, SIFT, PROVEAN, MutationTester, cuando fue posible. La patogenicidad de estas variantes se evaluó finalmente de acuerdo con el American College of Medical Genomics and Genetics (criterios ACMG).
RESULTADOS
La distribución por sexo incluyó 9 mujeres y 7 hombres. La edad media de los pacientes en el momento del diagnóstico de EII fue de 35 años. Y la edad media de aparición de síntomas gastrointestinales fue de 32 años.
Todos los pacientes habían tenido manejo con esteroide, sulfalazina o enemas al menos una vez en la vida. Once pacientes requirieron manejo farmacológico con terapia biológica (Adalimumab 2, Infliximab 9). Diez pacientes (62,5%) presentaron algún tipo de fracaso terapéutico con los tratamientos estándar y, por lo tanto, se clasificaron con enfermedad clínicamente grave. En seis pacientes se documentó falla terapéutica al manejo con esteroides, en cuatro a la terapia biológica (Adalimumab, Infliximab), y en un paciente falla terapéutica con Azatriopina. Tres de los dieciséis pacientes (18,75%) tenían antecedentes familiares positivos de EII.
Genotipificación
En cuanto a los resultados genéticos, todos los pacientes presentaron al menos un SNP relacionado con riesgo de EII en más de un gen. En Tabla 1 se resume los SNPs encontrados y su clasificación según American College of Medical Genetics and Genomics (ACMG).
Tabla 1 Genotipificación de los pacientes con EII. En las columnas se encuentran los genes evaluados. VUS: Variantes de significado clínico desconocido. Benigna: Variantes descritas sin asociación a la patología según la literatura y las bases de datos globales actuales.

Seis de dieciséis pacientes (37,5%) tenían los siguientes SNPs en el gen CD48: rs6667145 y rs2295615. Once de dieciséis pacientes (68,75%) tenían los siguientes SNP en el gen CD6: rs11230562, rs2074233, rs11230563. El último SNP se ha informado en pacientes con EII. Dos pacientes con CU y 1 paciente con EC tenían el SNP rs149214040 en el gen STAT3; este gen juega un papel patogénico en la colitis a través de la regulación de las células T.
Ocho de dieciséis pacientes tenían el SNP rs2304256 en el gen TYK2. En los japoneses, se ha informado que este SNP en combinación con diferentes SNP en el gen STAT3 aumenta la susceptibilidad a la EC, sin embargo, ninguno de nuestros pacientes con polimorfismos en TYK2 tenía polimorfismos en STAT3 al mismo tiempo.
El rs2228044 en el gen il6ST se encontró en 4 pacientes con EC y en 4 pacientes con CU. Las variantes de este gen se han asociado con la EII y el cáncer de colon, sin embargo, el SNP informado en nuestro paciente no se ha asociado antes con EII.
Tres pacientes diagnosticados clínicamente con EC, tenían variantes heterocigotas en NOD2: c.2104C> T (p.Arg702Trp), c.2107C> T (p.Arg703Cys), c.2722G> C (p.Gly908Arg), todos ellos tienen interpretaciones contradictorias de la patogenicidad (VUS vs. Probablemente benigno). Otro paciente con EC tenía la variante benigna c.2863G> A (p.Val955Ile) en el gen NOD2 (Tabla 1).
Al analizar la presencia de variantes genéticas se puede evidenciar que estuvieron presentes en al menos un paciente con CU, pero en cambio en los pacientes con EC hubo 5 genes ausentes (Tabla 2, Figuras 1 y 2). En este punto es importante analizar la significancia estos genes. También se observó que, en los pacientes con CU, los siguientes genes están presentes en más del 50% de los pacientes: CD48, CD6, TYK2, y en los pacientes con EC, los siguientes genes estuvieron presentes en más del 50% de los pacientes: CD6 e ITGAM (Figuras 1 y 2).
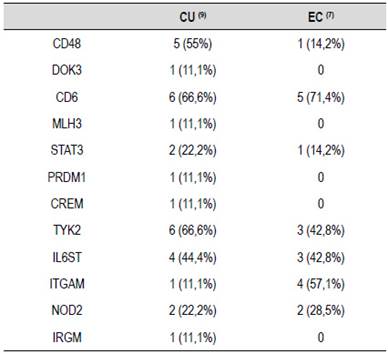
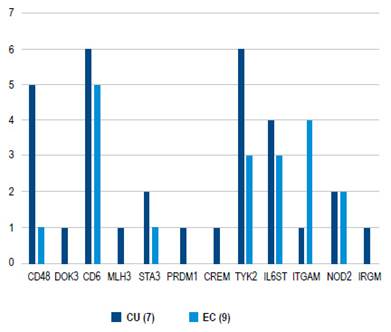
Figura 1 Variantes genéticas presentes en subtipos de enfermedad inflamatoria intestinal, según frecuencia.
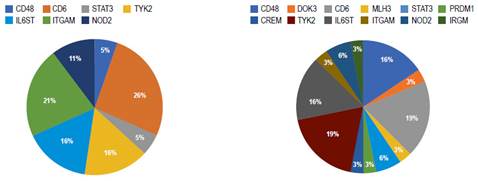
Figura 2 Comparación de variantes genéticas encontradas de acuerdo a subtipo de enfermedad inflamatoria intestinal.
En nuestra serie de casos los genes más frecuentes sin discriminación según patología fueron CD6 (22%), TYK2 (18%) y IL6ST (16%). El gen más frecuente fue el CD6 y no hay diferencias entre CU y EC ni en el promedio de otros genes comprometidos (Tabla 3).
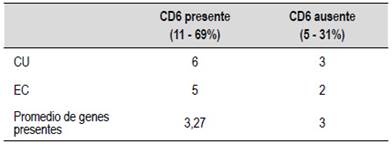
Análisis por número de genes expresados por pacientes
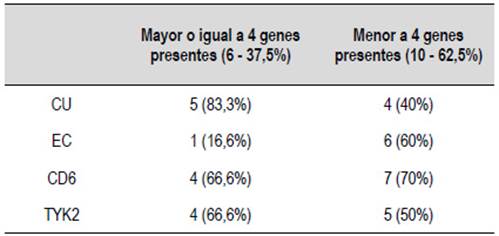
Se observó en tres pacientes la presencia de hasta 5 genes, 4 en tres pacientes, y en 5 pacientes tres de ellos (Tabla 4). Por otro lado, al comparar por número de genes comprometidos vemos que los pacientes que tienen más de 4 genes presentes el 83,3% son CU, en cambio en los que tienen menos de 4 genes presentes, el 60% son EC (Tabla 5).

DISCUSIÓN
La EII es una enfermedad multifactorial, con múltiples factores ambientales asociados incluyendo el microbioma gastrointestinal y que además incluye la predisposición genética como posible factor desencadenante 12. Los estudios de GWAS han identificado múltiples loci genéticos en el huésped que parecen mostrar una asociación significativa con la EII. Estos genes cumplen funciones en varias vías clave dentro de la inmunidad innata (por ejemplo, el gen NOD2), la señalización de citocinas (JAK2, STAT3, y TYK2) y el mantenimiento de la integridad de la barrera intestinal (HNF4A, CDH1 y MUC19) 25-27. En el presente estudio se encontró que la mayoría de los pacientes tenían una sumatoria de variantes clasificadas como benignas en los genes evaluados. En dos pacientes se encontraron tres variantes que la literatura ha reportado como de significado incierto (VUS por sus siglas en inglés, variants of unknown significance) 28. En el paciente (001) con diagnóstico clínico y anatomopatológico de CU se encontraron dos VUS en los genes DOK3 y MLH3.
Por otro lado, la actividad del gen DOK3, que codifica para una molécula transductora de receptores tirosinkinasa, ha demostrado regular negativamente el polímero JAK2/STAT3 la producción de citoquinas por los neutrófilos colónicos esto en modelos murinos, por lo que ha sido reportado hasta el momento como gen de susceptibilidad en uno de los estudios de asociación genómica. Este gen, aunque no es un actor epigenético, interactúa directamente con el gen STAT3 que al ser factor de transcripción es un modificador epigenético en la enfermedad inflamatoria intestinal 29. El gen DOK3 un regulador negativo de la detección de lipopolisacáridos (LPS) a través de la señalización de TLR4-ERK en macrófagos, regula la señalización de TLR3 a través de TRAF3-TBK1-IRF3 e influye en la secreción de interferón (IFN) -β31-35 30. Esto explicaría una posible alteración en la regulación inmune. Estudios en animales demuestran que la deficiencia de DOK3 promueve un desequilibrio en la microflora intestinal y por lo tanto una mayor susceptibilidad a la colitis 29. La susceptibilidad de DOK3 a la colitis parece explicarse mediante un mecanismo en el que la alteración de DOK3 restringe la señalización de las vías JAK2 / STAT3 de los neutrófilos del colon, lo que limita la expresión de la molécula S100a8 / 9, indispensable en el mantenimiento del microbioma intestinal comensal protectora, finalmente haciendo que se pierda la homeostasis intestinal 29. La variante en el DOK3, clasificada como VUS deberá seguirse en el tiempo para su reclasificación de acuerdo a los nuevos conocimientos científicos.
El gen NOD2 que por sus siglas corresponde a la proteína 2 que contiene el dominio de oligomerización de unión a nucleótidos confiere el mayor riesgo de EII. Es una proteína citoplasmática expresada esencialmente por células inmunitarias y células presentadoras de antígenos denotadas por linfocitos, macrófagos, células epiteliales (incluidas las células de Paneth ileales) y células dendríticas respectivamente. De hecho, se sabe que la activación de NOD2 provoca la liberación de varias moléculas proinflamatorias y antimicrobianas como IL1β, TNF-α, IL6, IL8 y α-defensinas. Esto desencadena, a su vez, el reclutamiento y la activación de diversas células inmunitarias innatas junto con la activación y diferenciación de células inmunitarias adaptativas 31. Del mismo modo, las mutaciones en NOD2 alteran la capacidad de las células de Paneth para reconocer y eliminar patógenos invasores, provocando el desarrollo de lesiones intestinales inflamatorias y disrregulación del microbiota por aumento de la concentración de bacterias huésped en el íleon. Una de las variantes más frecuentes, la G908R [rs2066845], fue encontrada en nuestra paciente 002, que según la literatura le confiere un riesgo entre dos y cuatro veces mayor de padecer EII (en particular EC) 8. Mucho se ha avanzado en la comprensión del gen NOD2 como molécula clave en la EII, incluso hay ensayos que pretenden disminuir la sintomatología de la EII mediante la activación o inhibición de NOD2 y de moléculas clave en su vía de señalización, convirtiéndolo en blanco terapéutico. Dado que NOD2 es una molécula clave en la EII, son muchas las investigaciones en torno a su posible aplicación clínica. Se han realizado estudios para aliviar la EII mediante la activación o inhibición de NOD2 y de moléculas clave en su vía de señalización 22.
El gen MLH3 codifica para una proteína que hace parte de la maquinaria de reparación de errores de apareamiento del ADN (MMR, Missmatch repair) y es importante debido a su papel en el mantenimiento de la integridad genómica y su conocida asociación con cáncer de colon hereditario no polipósico. Se ha descrito en la EII por su impacto en la represión epigenética en el proceso inflamatorio e hipóxico en modelos animales 31. Adicionalmente en estudios de GWAS se ha demostrado la posible asociación entre algunos SNPs en este gen y el desarrollo de CU 9,32. Lo cual coincide con nuestro estudio al encontrar una variante clasificada como de significado clínico incierto en un paciente con CU.
El haber encontrado dos VUS en un paciente con CU haría suponer que la presencia de múltiples loci en diferentes genes de susceptibilidad podría tener efectos aditivos contribuyendo a incrementar el riesgo de desarrollar EII, sin embargo, hasta el momento, son necesarios más estudios que permitan concluir el efecto aditivo de variantes en genes con funciones sobre el microbioma intestinal 11.
En el paciente 0004 con diagnóstico clínico y anatomopatológico de CU, se encontró una VUS en el gen PRDM1. Este gen codifica para una proteína que es un represor transcripcional que se une al ADN, ejerciendo sus funciones represivas mediante el reclutamiento de enzimas modificadoras de histonas. Diferentes estudios en la literatura han mostrado asociación significativa entre algunas variantes en este gen y el riesgo de desarrollar tanto CU como EC, por ejemplo la variante Ser354Asn, asociada significativamente con EC: odds ratio [OR] de 1,23 (95% CI=1,07-1,40), y las variantes rs7746082, rs6568421 y rs6911490 asociadas con un incrementos en el riesgo de desarrollar CU de hasta 8 veces más con respecto a población control 33. Aunque la variante en el gen PRDM1 de la paciente no ha sido previamente descrita en estudios de GWAS, también serían más estudios de asociación y funcionales para poder esclarecer el verdadero significado de la misma en la patogénesis de la enfermedad.
El paciente 14, con diagnóstico clínico y anatomopatológico de EC, presentó una variante con conflicto de patogenicidad según clinvar en el gen NOD2 (c.2026C>T, p.Arg703Cys (rs5743277)). Esta variante tiene una frecuencia alélica en gnomAD (Genome Aggregation Database) de 0,00283. Esta variante se ha descrito en la literatura en individuos con enfermedad autoinflamatoria 34,35, y también se ha descrito con mayor frecuencia en individuos con EC y CU 36,37. En conclusión, esta variante en el gen NOD2 pareciese que pudiese contribuir a la EC como factor de riesgo, posiblemente junto con otros factores genéticos y / o ambientales, pero son necesarios más estudios, porque hasta la fecha no hay evidencia suficiente para otorgar patogenicidad.
Algunas posibles limitaciones del estudio, incluyen que se trata de un estudio retrospectivo, con un número pequeño de pacientes, siendo esta una serie limitada de 16 pacientes. También, no haberse podido contar con una población control, por lo que no fue posible realizar un estudio de asociación de casos y controles. Por otro lado, el desarrollo del estudio en un solo centro podría limitar su validez externa: por ejemplo, la población atendida en este centro suele ser de un estatus socioeconómico medio a alto, lo que le confiere características particulares. Sin embargo, este estudio presenta múltiples ventajas, incluyendo que estos resultados demuestran la amplia variabilidad genética en pacientes colombianos con EII, donde más del 65% de los pacientes presentaban fenotipo clínicamente grave, y se espera que el seguimiento continuo de estos pacientes ser valioso, ya que las características clínicas entre los individuos son generalmente variable y diversa. Además, otras características, como las manifestaciones extraintestinales y otras complicaciones con compromiso multisistémico, afectan tanto el pronóstico a largo plazo como calidad de vida en pacientes con EII, destacando así la necesidad de recopilar una base de datos completa de fenotipos en EII para avanzar en el diagnóstico y tratamiento. La verificación por más de dos investigadores de los datos de los registros clínicos pudo además disminuir el sesgo de transcripción. En general, este estudio es el primero en informar las características clínicas de los pacientes colombianos con diferentes mutaciones, y estos hallazgos amplían los conocimientos fenotípicos y genotípicos en EII.
Aunque no hay una correlación fenotipo genotipo en la enfermedad inflamatoria intestinal si se ha reportado que hay una relación epistásica, es decir una relación aditiva, por ejemplo, entre los genes IBD5/NOD2, IL-10/IL10RB/HO1 y JAK1/TYK2/STAT3 lo cual está en sincronía con gravedad de la enfermedad y pobre respuesta al tratamiento 38. En nuestro estudio reportamos variantes en 2 de estos genes, las cuales han sido previamente reportadas en pacientes con EC.
En conclusión, consideramos que este estudio es una primera aproximación a la caracterización genotípica de pacientes con EII en población colombiana con resultados que indican la presencia de variantes en genes de predisposición a esta patología compleja, que, aunque sin patogenicidad confirmada al momento actual, parecen sugerir que la sumatoria de estas pueden contribuir en la fisiopatología de la enfermedad. Hemos podido demostrar que nuestra población es similar en cuanto a variantes en línea germinal que aumentan el riesgo o participan en una de las vías de señalización que se afectan en la enfermedad inflamatoria intestinal. Lo cual ubica a Colombia en un país a la vanguardia en temas de generación de conocimiento que permita avanzar en el conocimiento de esta patología con miras a mejorar la respuesta terapéutica e individualizar a los pacientes para dar una terapia dirigida. Son necesarios más estudios de genotipificación y estudios funcionales que permitan identificar biomarcadores o blancos terapéuticos para el desarrollo de nuevos fármacos. Además, generar conocimiento entre genes de susceptibilidad, cambios epigenéticos y las ciencias ómicas, lo cual permita enfoques confiables para la detección precoz de riesgo de enfermedades e interacciones entre patologías autoinmunes.

