











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
La Ataxia-telangiectasia (AT) es una enfermedad rara, con presentación multisistémica, que aún es de poco sospechada en la práctica clínica neurológica en regiones con poco a acceso a tecnologías de diagnóstico genómico como Perú y varios países de Latinoamérica. En esta revisión actualizada sobre AT compartimos información relevante sobre la epidemiología, manifestaciones clínicas, diagnóstico y tratamiento de AT. Asimismo, compartimos información obtenida por una búsqueda sistemática de casos de AT publicados en el Perú.
Definición y aspectos históricos
La AT es una enfermedad rara con afección multisistémica, caracterizada por ataxia cerebelosa progresiva y disartria, infecciones recurrentes, trastornos del movimiento, telangiectasias óculo-cutáneas, y riesgo de cáncer 1-3. La primera descripción de esta enfermedad fue realizada por la neuróloga belga Louis Bar en 1941, quien describió el caso de un niño de nueve años con marcha atáxica, hipotonía, nistagmo bilateral, discapacidad intelectual y telangiectasias en piel y conjuntivas 4, acuñando el epónimo de Síndrome de Louis-Bar. En 1958, se propuso la denominación de ataxia-telangiectasia a propósito de la descripción de ocho casos (5). Se han descrito casos de AT que asocian hallazgos clínico-patológicos variados como la hidromielia, bronquiectasia, fibrosis pulmonar, hipoplasia del timo, esclerodermia, hipogonadismo y disgerminoma, elevaciones séricas de alfa fetoproteína, así como formas atípicas con ausencia de telangiectasias (6-8). En 1995, se identificó que variantes patogénicas en el gen ATM (ataxia-telangiectasia mutated) son responsables del fenotipo AT 9. En 2015, se ha propuesto la denominación de síndrome ATM para explicar mejor la expresión sistémica y variable de síntomas asociados a variantes genéticas del gen ATM 10.
Etiopatogenia
La AT es una enfermedad autosómica recesiva originada por variantes en el gen ATM (11q22-q23) que conlleva a una expresión anormal de la proteína ATM (11, 12). Esta una quinasa serina/treonina involucrada en el reconocimiento y la respuesta celular al daño de ADN por radiación ionizante en las roturas de doble hebra en el ADN 13). En condiciones fisiológicas, cualquier daño en el ADN activa la proteína ATM, y esta, a su vez, activa una cascada de proteínas responsable de la reparación a través de la vía de recombinación homóloga o la detención/apoptosis de la célula dañada (14, 15). La afectación en la expresión en diversos órganos y sistemas origina el fenotipo AT 2).
Hasta noviembre de 2022, se han identificado más de 5 mil variantes (https://databases.lovd.nl/shared/genes/ATM), incluyendo variantes sin sentido “nonsense”, cambio de sentido “missense” y variantes en el sitio de corte y empalme “splicing variants”, algunas de las cuales conservan parcialmente la actividad quinasa de la proteína ATM 12). Al ser una enfermedad recesiva, las variantes causales de AT pueden presentarse en homocigosidad o en heterocigosidad compuesta 16). Variantes que no truncan la formación de la proteína estarían asociadas a formas atípicas de AT 2).
Epidemiología
AT es la segunda ataxia congénita más frecuente a nivel mundial, con una prevalencia variable entre 1: 40 000 a 1: 300 000 17). La AT representaría hasta el 5,2% de causas de inmunodeficiencias primarias a nivel mundial 18).
AT en Perú
En el Perú, la AT está descrita en pocos reportes y series de casos. Realizamos una búsqueda sistemática según guía PRISMA (figura 1) de las bases de datos LILACs/Scielo, Medline/PubMed y Scopus con fecha de corte hasta el 30 de noviembre de 2022. La estrategia de búsqueda consistió en palabras clave como "Ataxia Telangiectasia", “Ataxia Telangiectasia Syndrome”, y otros términos relacionados tales como “Cerebello-Oculocutaneous Telangiectasia” y “Louis-Bar Syndrome”, restringiendo la búsqueda a Perú. Los criterios de inclusión fueron: 1) reportes de casos o series de casos de ataxia telangiectasia en Perú; 2) publicaciones que describieron datos sociodemográficos y clínicos de los pacientes (incluyendo datos individuales o agregados) y 3) disponibilidad del texto completo o resúmenes en inglés, español o portugués. Se utilizó el programa Rayyan para seleccionar los estudios y eliminar duplicados. Posteriormente se seleccionaron los reportes y extrajeron los datos en Microsoft Excel. Los datos extraídos incluyen: nombre del primer autor, año de publicación, sexo, edad de inicio, cuadro clínico e información relacionada al estudio genético. Se realizó un enfoque narrativo para sintetizar los estudios incluidos.
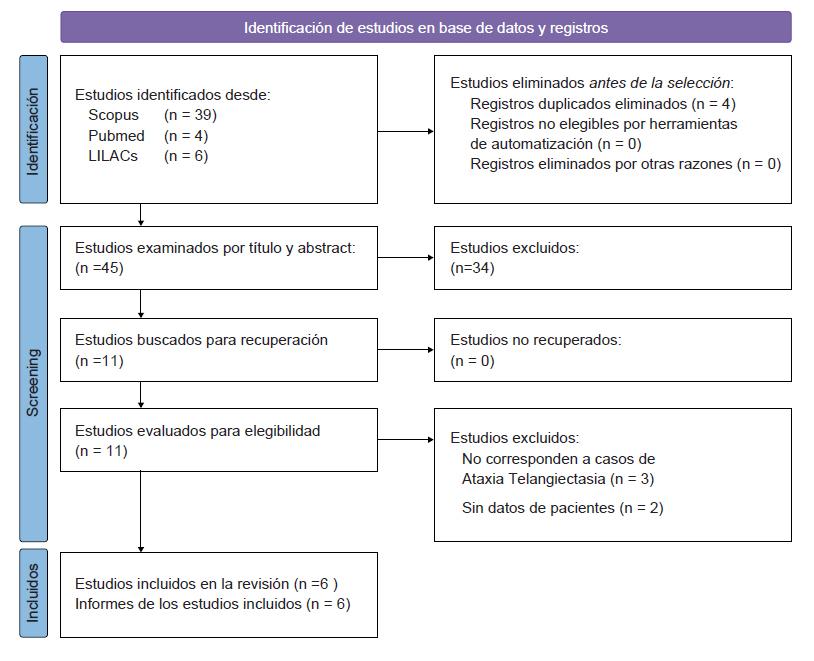
Se encontraron 4 artículos en PubMed/Medline, 39 artículos en Scopus y seis artículos en LILACs/Scielo. Del total, se descartaron 43 estudios que no correspondían a casos de Ataxia-telangiectasia y duplicados, identificando un total de seis estudios. Luego de la extracción de información individual y familiar, se identificaron 21 casos con diagnóstico de Ataxia-Telangiectasia, 10 de los cuales cuentan con descripción clínica asociada (19-24). Solo tres de los 21 casos descritos describen estudios genéticos, con variantes en heterocigosis compuesta en el gen ATM. El listado completo de individuos AT identfificados y principales características se listan en la tabla 1 y figura 2.
Tabla 1: Casos de Ataxia-Telangiectasia reportados en Perú
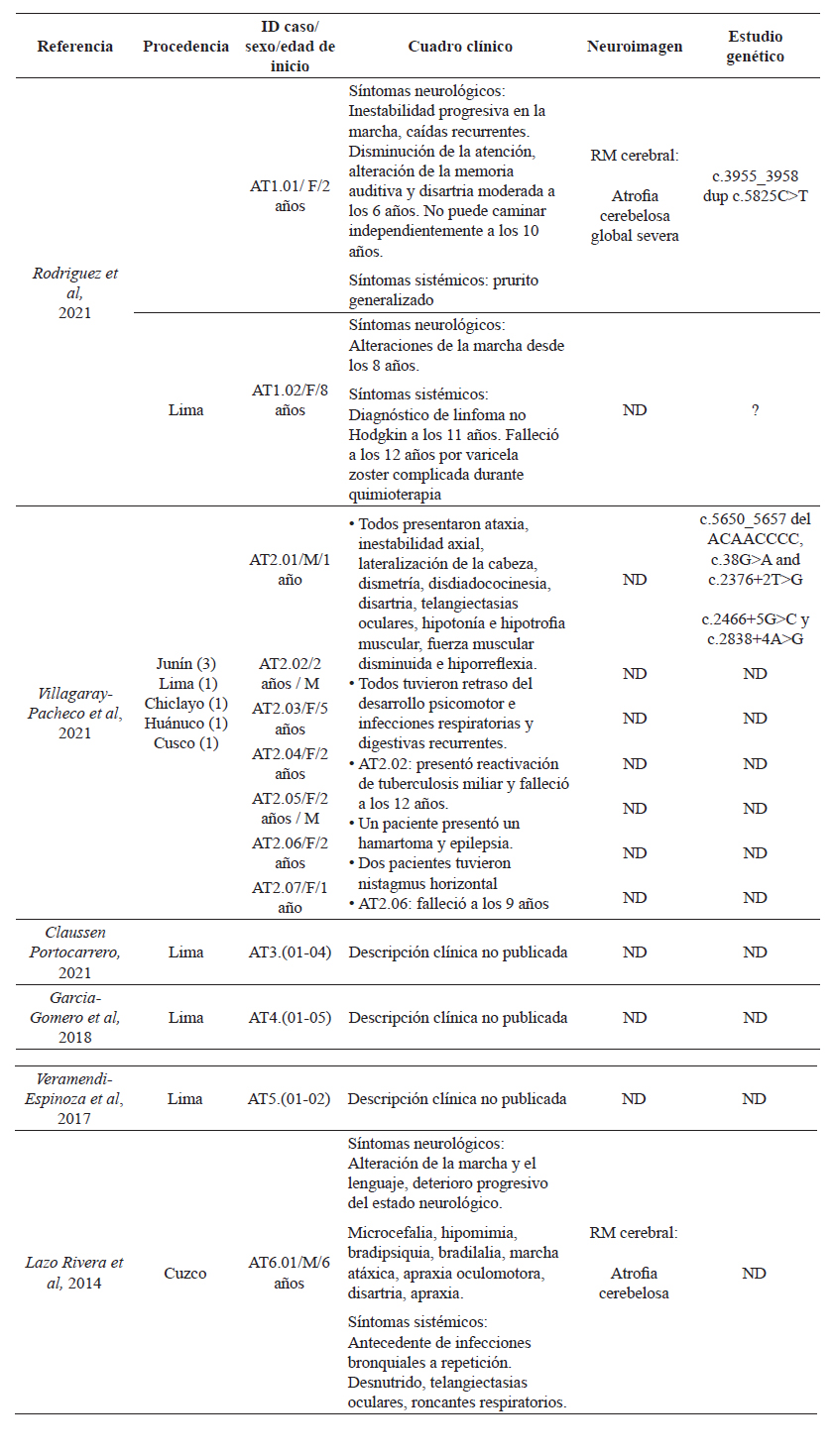
ND. No disponible. RM: resonancia magnética
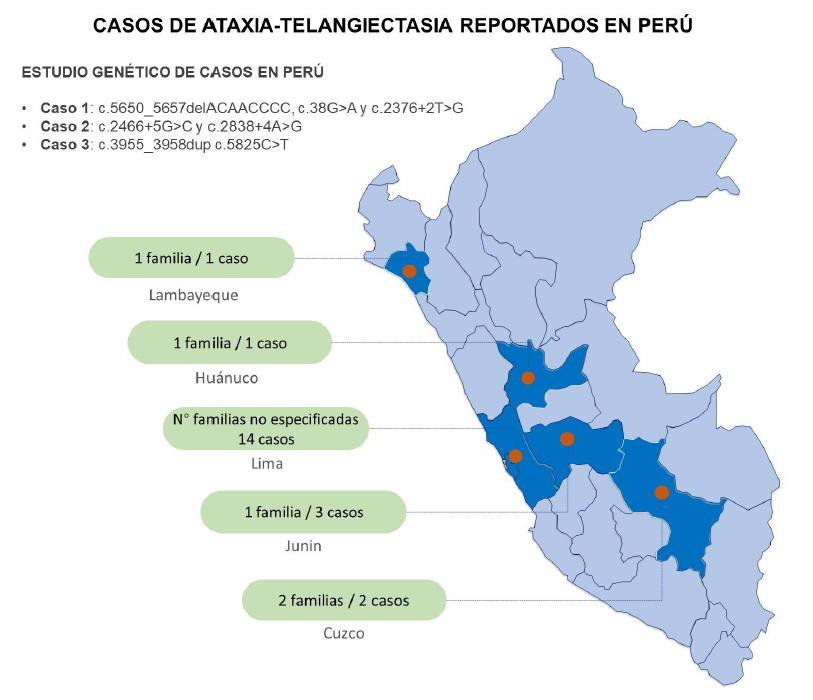
Manifestaciones clínicas
Las manifestaciones clínicas suelen empezar en la infancia e incluyen la ataxia cerebelosa, apraxia oculomotora, corea y disfunción cognitiva 2). La afectación multisistémica comprende inmunodeficiencia, endocrinopatía, radiosensibilidad, inestabilidad cromosómica y telangiectasia óculo-cutáneas. También se asocia con una alta incidencia de cáncer, predominantemente leucemia y linfoma. En los casos reportados en Perú, la edad de inicio fue de 3,1 años, con un intervalo de 1 a 8 años. La mayoría de ellos cursó con ataxia y síntomas cerebelosos, telangiectasias oculares, hipotonía e hipotrofia muscular. Los signos y síntomas de la AT y sus frecuencias relativas se resumen en la tabla 2.
Tabla 2 Frecuencia relativa de las manifestaciones clínicas de Ataxia-Telangiectasia.
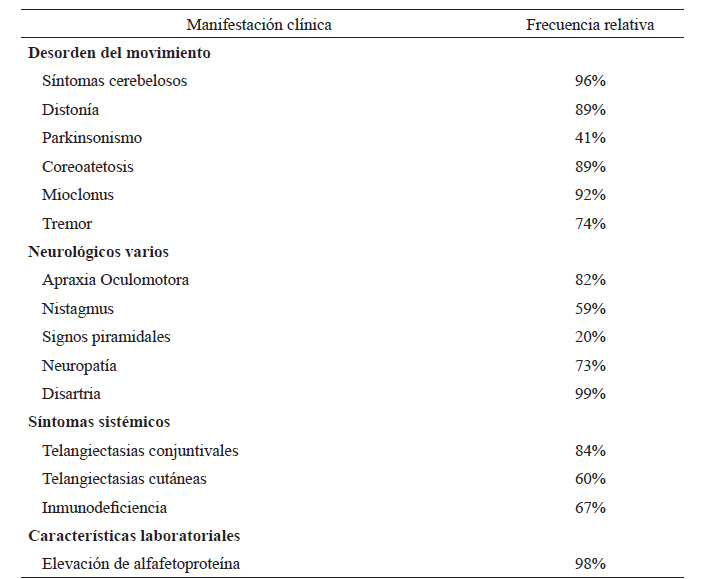
Adaptado y traducido de Levy A & Lang (2018) (25).
Tabla 3 Diagnóstico diferencial de ATM
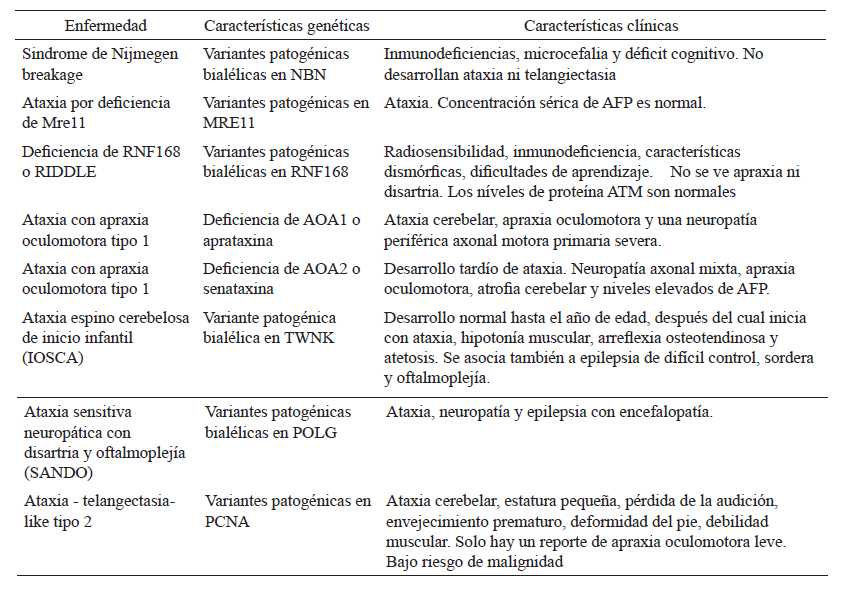
AFP: alfafetoproteína, ATM: gen de Ataxia-Telangiectasia. Adaptado y traducido de Raslan et al (2021); Moreira et al (2018) (55, 56)
Ataxia cerebelosa. Es la manifestación clínica más frecuente de la AT clásica. La ataxia axial suele ser la manifestación inicial y aparece en el curso de la enfermedad hasta en 96% de casos 25). La edad de inicio promedio es de 1,4 años con una progresión lenta en 2 a 5 años y estabilización posterior 26). Habitualmente el patrón de la marcha suele presentarse con menor ampliación de la base de sustentación de lo esperado, debido a la mezcla de ataxia y apraxia para la marcha 26). La ataxia segmentaria suele aparecer aproximadamente cinco años después del inicio de la ataxia axial 27). En las formas de inicio tardío, la ataxia es menos frecuente (28).
Telangiectasias óculo-cutáneas. Las telangiectasias oculares, que son características, mas no patognomónicas, aparecen en promedio a los 6 años, pero pueden presentarse desde el nacimiento hasta los 15 años 26). Las telangiectasias en AT se han descrito en una frecuencia variable de 14,48-84% de los casos (29). Las telangiectasias oculares pueden estar ausentes hasta en un 50% de casos de AT, especialmente en las formas atípicas 29). Las cutáneas, se localizan frecuentemente en el lóbulo de la oreja, las superficies malares de la cara, el dorso de las manos, las fosas poplíteas, la región antecubital, el cuello y el área esternal 27).
Trastornos del movimiento. La AT puede asociar una gran variedad de movimientos anormales, como la coreoatetosis (89% de casos), distonía (89%), mioclonía (92%), temblor de acción (74%) y parkinsonismo (41%) (25). Se han reportado coreoatetosis axial, segmentaria y generalizada 26 Las distonías descritas incluyen de tipo focal, segmentaria y generalizada (30, 31). El temblor de acción es el más frecuentemente reportado, seguido por el temblor de reposo (31).
Movimientos oculares anormales. Han sido reportados en alrededor de 82% 25). Se manifiestan principalmente, como apraxia oculomotora en un 82%, alteración de movimientos sacádicos (77%) y de seguimiento (75%) (25, 32, 33). Los movimientos sacádicos suelen presentarse a inicios de la enfermedad y pueden progresar a una apraxia oculomotora u oftalmoplejía 25). Además, se han descrito alteraciones en las fijaciones de la mirada con presencia tanto de intrusismo sacádico como de nistagmo 34). Hasta en el 59% se ha encontrado nistagmo espasmódico, evocado por la mirada, pendular, de rebote, nistagmo horizontal en la mirada primaria y lateral, nistagmo post rotacional, y nistagmo alternante periódico (2, 35). También se han descrito defectos en el reflejo vestíbulo-ocular y estrabismo (2, 3, 33).
Cognición. El compromiso cognitivo no suele ser evidente al inicio de la enfermedad, manifestándose a lo largo de los años como un deterioro cognitivo leve a moderado en más de un 50% de casos (25, 36). Dunn et al reportaron deterioro cognitivo en aproximadamente un tercio de los pacientes; sin embargo, estas cifras podrían variar según el tiempo de vida debido que progresa según avanza la enfermedad (37, 38). Los dominios cognitivos alterados son la atención, memoria no verbal, fluencia verbal, percepción de intervalos de tiempo, pensamiento abstracto, cálculo y la función ejecutiva (2, 39)
Neuropatía periférica. Se asocia polineuropatía axonal sensitiva y motora luego de aproximadamente 10 años del inicio de síntomas, manifestados principalmente con arreflexia osteotendinosa, contracturas de miembros superiores e inferiores y deformaciones en los pies como pie cavo 25). El signo de Romberg es negativo e, una diferencia clínica relevante para el diagnóstico diferencial con otros fenotipos como la ataxia de Friedreich 40).
Inmunosupresión. La disfunción de la proteína ATM está asociada a estados variables de inmunosupresión que se presentan hasta en un 67% de pacientes 41). La inmunodeficiencia predominante es de tipo celular, seguida de hasta un 10% de casos con inmunodeficiencia humoral o mixta. (2, 42). Las formas más severas de inmunodeficiencia se han descrito a edades más tempranas de la enfermedad (2, 25). El patrón de inmunodeficiencia incluye disminución de inmunoglobulinas IgG, IgA, o IgM (50-80%) y linfopenia a predominio de linfocitos T, que se asocian a infecciones recurrentes e incremento de morbimortalidad (2, 25, 36, 43). Ocho de los 21 casos descritos en Peru presentaron infecciones a repetición, principalmente respiratorias, probablemente secundarias a inmunosupresión.
Enfermedad pulmonar. Las enfermedades respiratorias representan la segunda causa de muerte en AT (3). El compromiso pulmonar se desencadena por la coexistencia de inmunodeficiencia celular, infecciones sinopulmonares que van incrementándose con la edad, estrés oxidativo y fenómenos de inflamación sistémica crónica (25, 42). En algunos casos, se han descrito cambios compatibles con enfermedad pulmonar obstructiva crónica 44).
Predisposición al cáncer. La aparición de neoplasias malignas son la principal causa de muerte en AT, presentándose hasta en un 22% (3, 25). La etiopatogenia del cáncer en AT estaría en relación con la alteración en síntesis de ADN y una mayor susceptibilidad a radiaciones ionizantes que conllevan al estrés genotóxico, inestabilidad genómica y cáncer (2 45). Las neoplasias más comunes en pacientes menores de 20 años son la leucemia y el linfoma (36, 42, 46). En adultos, otros tipos de neoplasias se evidencian, como el de mama, gástrico, hepático, glándulas parótidas, y esofágico (2, 47). Los individuos portadores de variantes patogénicas en el gen ATM, habitualmente familiares de primer o segundo grado de pacientes, tienen riesgo incrementado de neoplasias y se recomienda evaluaciones preventivas regulares (45, 47). En los casos reportados en Perú se describe un caso de linfoma no Hodgkin y un caso de Hamartoma.
Manifestaciones endocrinológicas. Las alteraciones endocrinas en AT son variadas, pero poco frecuentes. Se han descrito casos de deficiencia de vitamina D, dislipidemia, disminución de sensibilidad a la insulina, diabetes y atrofia gonadal y excepcionalmente disfunción tiroidea (2, 3, 36, 42, 48).
Hallazgos en neuroimagen. Los principales hallazgos observados con la resonancia magnética son atrofia cerebelar del vermis, pedúnculo cerebelares medio y superior, lesiones a nivel del tálamo, desmielinización en ambos núcleos basales y dilatación ventricular (49, 50). Los cambios imagenológicos suelen ser muy sutiles durante los primeros 10 años de enfermedad 49). A partir de los 40 años, es posible encontrar otros hallazgos como depósitos de calcificaciones en el tronco cerebral y a nivel subcortical además de lesiones ocupantes de espacio en sustancia blanca del lóbulo frontal derecho 50).
Diagnóstico
Se debe sospechar AT en todo paciente con ataxia progresiva con o sin telangiectasias, trastornos del movimiento, apraxia oculomotora, infecciones recurrentes con o sin antecedente familiar 51). Los exámenes auxiliares de mayor utilidad incluyen la resonancia magnética que demuestra hipoplasia cerebelosa de vermis y de ambos hemisferios cerebelosos, la AFP sérica elevada mayor a 10ng/ml; en caso de inmunodeficiencia asociada son de utilidad dosaje de inmunoglobinas séricas 52). El diagnóstico definitivo se establece con una prueba genética (de gen único, panel multigen, exoma o genoma) para identificar variantes patogénicas bialélicas del gen de la ATM 53). Las técnicas de inmunoblotting pueden ser usadas como herramienta diagnóstica cuando se identifica una sola variante patogénica 54).
Diagnóstico diferencial.
Dentro de las patologías que se pueden confundir con AT, encontramos la enfermedad AT-like tipo 2, síndrome RIDDLE, ataxia con apraxia oculomotora tipo 1 y 2, ataxia de Friedreich y ataxia espinocerebelosa con neuropatía axonal (Tabla 2)
Manejo
Actualmente, la AT no tiene tratamiento curativo o modificador disponible. El manejo de AT debe ser integral y multidisciplinario. Incluye medidas farmacológicas para controlar algunos síntomas no farmacológicas como terapia física y ocupacional, asesoría nutricional, asesoría genética personal y familiar.
Farmacológico. Hay algunos estudios que proponen uso de amantadina, buspirona y fluoxetina para la coordinación, equilibrio y disartria (nivel de evidencia II-2). El temblor de intención puede ser tratado con clonazepam y gabapentina (nivel de evidencia II-2) 53). Se ha observado un alivio temporal de la ataxia cerebelosa con el uso de esteroides (nivel de evidencia II-1); sin embargo, debido a sus efectos adversos, se recomienda evaluar individualmente el riesgo-beneficio 41). En el caso de los desórdenes del movimiento como la corea y el parkinsonismo, se han utilizado agonistas dopaminérgicos y fármacos anticolinérgicos 53 La distonía segmentaria responde bien a la toxina botulínica (nivel de evidencia II-2) 41). Se ha descrito la utilidad de clonazepam y levetiracetam para el tratamiento de las mioclonías en ATM (nivel de evidencia II-3) 57). El tratamiento de las neoplasias en AT no tiene un protocolo de tratamiento estandarizado, se recomienda iniciar con dosis baja de quimioterapia y ajustar la dosis en forma individualizada 41). En caso de inmunodeficiencia e infecciones recurrentes considerar el uso de inmunoglobulina IV o SC.
No farmacológico. La implementación regular de terapia física, ocupacional y de lenguaje, contribuyen a disminuir la pérdida de función neurológica en AT con resultados positivos sobre la calidad de vida (2, 44). La Sociedad Europea Respiratoria (European Respiratory Society) sugiere exámenes de evaluación pulmonar y radiografías de tórax, para diagnósticos oportunos de complicaciones respiratorias. Se recomienda el uso de sonda nasogástrica y educar a los familiares en su uso y en precauciones durante la ingesta de alimentos para prevenir neumonías aspirativas por la disfagia progresiva (2, 51). Se recomienda vacunación contra los principales patógenos respiratorios como el haemophilus influenzae, virus de la influenza y neumococo 52).
Asesoramiento genético. Los padres de un individuo afectado son heterocigotos obligados. Los heterocigotos (portadores) de variantes de AT tiene riesgo incrementado de cáncer y enfermedad coronaria 2). Cada hermano de un individuo afectado tiene de 25% de estar también afectado, 50% de riesgo de ser portador asintomático y 25% de no ser portador. La mayoría de los individuos afectados con AT no tienen descendencia, los hijos de pacientes con AT son excepcionales (58). Los tíos paternos y maternos de un individuo AT tienen 50% de riesgo de ser portadores de variantes patogénicas del gen ATM. Se recomienda estudio genético a los familiares en riesgo, luego de la identificación de las variantes genéticas causales de AT en el probando.
Conclusiones y Perspectivas
La AT es una enfermedad rara o huérfana de compromiso sistémico que; además de la ataxia progresiva, la apraxia oculomotora y las telangiectasias óculo-cutáneas; asocia estados de inmunodeficiencia con infecciones recurrentes y mayor riesgo de cáncer. El diagnóstico precoz de las personas afectadas por AT, así como de los familiares en riesgo, permite no solo la implementación de un manejo multidisciplinario con enfoque de control de síntomas y terapia integral, sino también con enfoque de prevención de complicaciones, y vigilancia de riesgo de cáncer en el probando y familiares en riesgo.
Los pocos casos de AT publicados en el Perú muestran las actuales limitaciones de nuestro sistema de salud para el diagnóstico y el manejo oportunos de la AT. La implementación de redes de colaboración interinstitucional, así como convenios intercambio prestacional entre los subsistemas de salud, permitirán realizar el diagnóstico genético oportuno de los casos de AT. El trabajo coordinado con grupos de pacientes y asociaciones de familias ayudaría a mejorar las coberturas de atención de pacientes con AT en el país. Garantizar la continuidad de la prestación de servicios de salud a pacientes pediátricos con AT que llegan a la adultez evita que se interrumpa el tratamiento instalado y se dé continuidad al manejo integral en cada caso. El acceso universal a tecnologías sanitarias como la inmunoglobulina en pacientes con AT disminuiría las infecciones recurrentes mejorando la calidad de vida de estos pacientes. La implementación de programas de prevención de cáncer para pacientes AT y familiares en riesgo en todos los subsistemas de salud, garantizaría la detección precoz y tratamiento oportuno esta complicación.
Finalmente, la implementación de estrategias multisectoriales que apoyen y promuevan la investigación clínica desde instituciones de salud como centros de salud, hospitales e institutos especializados, brindaría información local que facilite la toma de decisiones en salud en favor de los pacientes con AT y otras enfermedades raras y huérfanas.

