











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO  uBio
uBio 
 Permalink
Permalink
INTRODUCCIÓN
Las interacciones medicamentosas son el resultado de la administración conjunta de dos o más medicamentos. La forma en que estas se presentan puede llevar a alteración en la farmacodinamia y/o la farmacocinética de los fármacos. El resultado de este fenómeno genera cambios en la biodisponibilidad de los agentes terapéuticos, aumentándola o disminuyéndola. También se puede observar efectos que potencien o antagonicen los mecanismos de acción de los diferentes principios activos (Kennedy et al., 2016).
Un campo poco explorado y que acompaña el fenómeno de las interacciones es la farmacogenética; ciencia que busca describir las bases genéticas que causan la variación de la respuesta a los fármacos por parte de los individuos (Pirmohamed, 2016). La repercusión de la variabilidad genética entre sujetos viene a modificar tanto la farmacodinamia como la farmacocinética de los principios activos. Los sistemas enzimáticos CYP450, los sistemas de conjugación y algunos transportadores, como la glicoproteína P, son de los elementos más estudiados y representan uno de los mejores acercamientos a esta área.
En la actualidad, una práctica que se vuelve cada vez más común por parte de prescriptores es la polifarmacia, que es el uso simultáneo o excesivo de varios medicamentos (Castro-Rodriguez et al., 2016). Esta práctica puede variar según los patrones de prescripción asociados a una región, y puede ser influenciada por determinantes sociales, culturales, económicos y/o promocionales. Existe un rezago en el conocimiento de la farmacogenética de especies caninas y es necesario indagar más e integrar la información que se posee sobre las interacciones medicamentosas, la farmacogenética y la polifarmacia, debido a que solo así se podrá avanzar correctamente hacia una práctica donde se permita individualizar las terapias farmacológicas en las diferentes razas, así como de cada paciente tratado.
El objetivo de esta revisión es analizar el estado actual del conocimiento sobre la variación de la respuesta a los fármacos en la especie canina, tomando en cuenta las interacciones medicamentosas, producto de la polifarmacia, y el cómo la farmacogenética asume un papel de importancia para comprender estos fenómenos en las diferentes razas.
Farmacogenética
Este es un campo de estudio que busca relacionar las variaciones en el genoma y sus implicaciones en las respuestas farmacológicas que un sujeto puede tener a un fármaco. El término fue acuñado en la década de 1950, al observar anemias hemolíticas en algunos sujetos que tomaban tratamiento antimalárico (Ferraldeschi y Newman, 2011). Con el crecimiento del arsenal terapéutico y el uso simultáneo de diferentes drogas para uso en un paciente, se pone en evidencia la necesidad de una mayor atención a esta área científica debido a las variaciones en la biodisponibilidad y las posibles reacciones adversas que pueden acontecer. Entendiéndose como reacción adversa cualquier situación en la que se exacerbe o disminuya el efecto de una droga, así como también la aparición de nuevas respuestas no descritas en la media poblacional y ajenas a su efecto principal aprobado para una condición especifica, y donde se experimente una respuesta nociva y no intencionada (Coleman y Pontrefract, 2016; Montané y Santesmases, 2020).
En general las variaciones genéticas de importancia suelen ser características de razas específicas, entendiendo por raza el conjunto de organismos que poseen ancestros en común y una serie de características distinguibles que se han transmitido por herencia genética (Freishcher et al., 2008). Dentro de las variaciones heredables se suele dar importancia desde el abordaje farmacogenético, a todas aquellas que aparezcan en más del 1% en una población; y estas comúnmente suelen atribuirse a polimorfismos simples de un solo nucleótido (SNP, por sus siglas en ingles single nucleotide polimorphism) (Kongara, 2017). Al presentarse un cambio polimórfico en un gen las características del producto pueden variar en función del sitio o los sitios donde ocurran, pudiendo presentarse expresiones a nivel de proteína en donde su función no cambia; algunas son hiperreactivas o hipoactivas, así como también podrían manifestar secuencias de parada prematuras o tardías, que expresan proteínas afuncionales (Ramírez-Bello et al., 2013). A pesar de que ya se han descrito varios tipos de mutaciones polimórficas en medicina veterinaria, este sigue siendo un campo incipiente y de poco desarrollo en esta ciencia.
Generalidades de Respuesta a Opioides en Perros
Los cambios a nivel farmacocinético más notables son los que sufren los opioides, cuyo mecanismo de acción está principalmente asociado a su acción sobre receptores nociceptivos. Un ejemplo de esto sucede con el sulfato de morfina, que presenta una disminución en su biodisponibilidad al ser administrada por vía oral en perros de raza Beagle (Kongara, 2017). Las variaciones polimórficas de los sistemas enzimáticos encargados del metabolismo de opioides no se han estudiado a profundidad (Court, 2013). Existen reportes de polimorfismos a nivel del gen que codifica para el receptor opioide µ en perros, mientras que los datos para los receptores κ y γ son limitados (Kongara, 2017). Las mutaciones son el resultado de dos SNPs (C-15A y C207T) dentro del exón 1 para el gen OPRM1 (Hawley y Wetmore, 2010). El polimorfismo C-15A se encuentra principalmente presente en el Malamute de Alaska, Husky Siberiano y Labrador Retriever (Kongara, 2017). Las mutaciones de este tipo se asocian a problemas de unión entre droga-receptor. Para el caso ya mencionado, en la clínica se ha visto problemas de disforia en las razas ya comentadas (Hawley y Wetmore, 2010; Kongara, 2017). En el Cuadro 1 se presenta un resumen de reportes sobre el uso de opioides en perros, siendo estos considerados como la primera línea en el manejo de dolor agudo.
Cuadro 1 Aspectos generales sobre las diferencias asociadas a distintos tipos de opioides de uso veterinario

UGT: UDP-glucoroniltransferasa
Elaboración propia
Existen reportes de mutación en el receptor 1 de melanocortina (MC1R, por sus siglas en inglés) en humanos y ratones, donde se ha mostrado un aumento de la nocicepción en aquellos sujetos que la expresan (Liem et al., 2004; Kongara, 2017), y se asocia este fenómeno a la baja unión entre la hormona estimulante de melanocitos α (MSH-α, por sus siglas en ingles) y el receptor (Kongara, 2017). Esta condición aumenta los niveles MSH-α, lo que a la larga genera un retrocontrol negativo sobre su producción, y al estar está ligada al péptido opioide endógeno, a través de su precursor proopiomelanocortina; no se da la producción ni de uno ni del otro (Getting, 2006). La mutación de este receptor también se relaciona con diferencias en la coloración del pelaje de los sujetos que la presentan, dado que la variación de este receptor genera una pigmentación del cabello más clara en quienes la expresan (Kongara, 2017). La mutación del MC1R ha sido identificada en varias especies, incluyendo los perros, pero en estos últimos no se dispone de estudios sobre sensibilidad al dolor (Kerns et al., 2004).
Mutación del Gen MDR1
La primera descripción registrada de la glicoproteína P (P-gp, por sus siglas en inglés) como producto del gen MDR1 (por su nombre en inglés, multi-drug resistance), también conocido como ABCB1, fue realizada por Juliano y Ling (1976), y su presencia se evidenció en el ovario del hámster chino (Cricetulus griseus), mostrándose la resistencia que existía a la colchicina (Juliano y Ling, 1976; Correa y Castaño, 2014). El cambio de nombre de MDR1 a ABCB1 fue propuesto para que este gen se mantuviese acorde a la nomenclatura sistémica de la familia de transportadores ABC (ATP-binding cassete, por sus siglas en inglés) (Dean, 2005; Klintzsch et al., 2010). Por otro lado, la superfamilia de transportadores ABC son un grupo de transportadores que se caracterizan por ser dependientes de ATP (Mealey, 2013).
El principal interés que se tiene sobre este transportador gira en torno a distintos efectos terapéuticos que se observan en los sujetos que presentan mutación en el gen ABCB1 y por consiguiente los cambios manifiestos en la expresión de la G-gp (Mealey, 2008). La G-gp se expresa en células epiteliales del tracto intestinal, los túbulos contorneados proximales del riñón y hepatocitos, así como también a nivel de la barrera hematoencefálica en los capilares cerebrales (Mealey, 2013); lo que lleva a que la variación de este transportador provoque como resultado cambios en la biodisponibilidad de las drogas en los primeros casos mencionados y efectos acentuados a nivel del sistema nervioso central para el último (Fromm, 2000; Klintzsch et al., 2010).
La mutación nt230 [del4] del gen ABCB1 canino es la que se asocia al fenómeno que ocurre en la G-gp, y fue relacionada en un inicio a los fenotipos sensibles a ivermectina que se observaron en perros de raza Collie (Klintzsch et al., 2010), y que posteriormente se reportaron en otras razas en una frecuencia menor (Cuadro 2) (Monobe et al., 2015). La primera intoxicación por ivermectina en perros de raza Collie fue descrita en 1983 (Hugnet et al., 2004) y, posteriormente en 1987 se observó que no todos los Collie sufrían los síntomas de neurotoxicidad que se relacionan a esta droga (Correa-Salgado y Castaño, 2014). Los perros que expresan la mutación nt230 [del4] de forma homocigota presentan una mayor predisposición a las intoxicaciones por sustratos de la P-gp, mientras que aquellos que la presentan de tipo heterocigoto tienen menos predisposición y los que son homocigotos no mutantes (nativos) no presentan una susceptibilidad relevante (Monobe et al., 2015; Deshpande et al., 2016). A consecuencia de esto se han desarrollado una diversidad de técnicas moleculares, como la PCR, para identificar la presencia del gen mutante en los animales, ya que no en todos los casos se requiere evitar o reducir la dosis de los fármacos que son sustratos de este transportador (Klintzsch et al., 2010; Monobe et al., 2015).
Cuadro 2 Frecuencia aproximada de mutación del gen ABCB1 en razas caninas según mutación homocigota o heterocigota
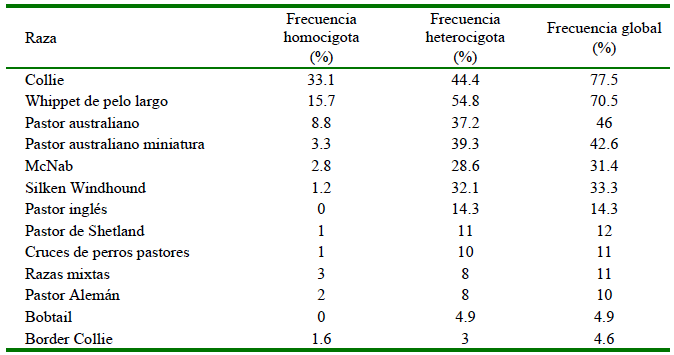
Elaboración propia, basado en datos de: Neff et al., 2004; Mealey y Meurs, 2008; Dekel et al., 2017
El uso de fármacos inhibidores de Ggp, como el antifúngico ketoconazol o el insecticida spinosad, se ha relacionado con un aumento de la toxicidad de los sustratos de este transportador, tal como ocurre con la ivermectina (Coelho et al., 2009; Schrickx, 2014). Debido a fenómenos como este se puede evidenciar el efecto de la polifarmacia en conjunto a las variantes farmacogenéticas. En el Cuadro 3 se presentan los fármacos que son sustrato de la G-gp y que deben ser evitados o se les debe reducir la dosis en caso de existir presencia de mutación del gen ABCB1.
Cuadro 3 Fármacos que son sustrato del transportador G-gp, según su categoría farmacológica

Fuente: Mealey y Fidel, 2015
Mutación del Gen Codificante para el Receptor Adrenérgico β 1
Aunque se dispone de escasos estudios, se ha logrado observar que existen dos sitios polimórficos a nivel de la cola del receptor adrenérgico β1 (Maran et al., 2013). Este polimorfismo fue identificado al estudiar razas caninas (Cavalier King Charles Spaniel, Terranova, Boxer, Gran Danés y Doberman Pincher) de alta incidencia de enfermedades cardiacas (Mealey et al., 2019). Los perros que presentan una doble deleción en el gen llegan a tener una menor respuesta a los fármacos como el atenolol (Meurs et al., 2015).
Variaciones en el Metabolismo de Drogas
Las reacciones metabólicas, tanto en los caninos como en el humano se agrupan en una fase I, que comúnmente es de origen microsomal y donde participan los sistemas enzimáticos del CYP450, y una fase II, también conocida como fase de conjugación, donde diversos sistemas enzimáticos ayudan a modificar de los xenobióticos a través de la agregación de conjugados que permiten incrementar de manera más efectiva la polaridad y el tamaño de las moléculas, y así favorecer la excreción renal o biliar de aquellas donde la fase Ino logró ese objetivo (Trepanier, 2006; Lemke et al., 2013). Es importante mencionar que estos procesos no tienen que ocurrir en todas las drogas. Asimismo, una fase es independiente de la otra, pudiendo ocurrir una de las dos o ambas si así fuera necesario (Figura 1).
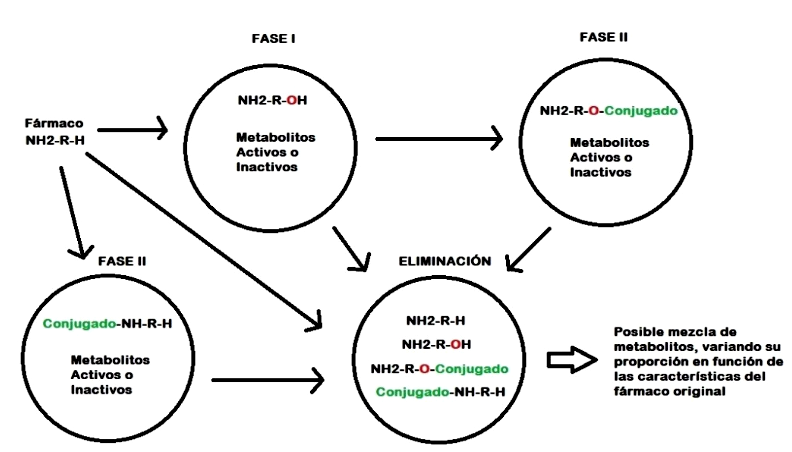
Figura 1 Posibles rutas del metabolismo de un fármaco según sus características químicas originales.
Los sistemas CYP se agrupan en familias (número), subfamilias (letra) y se identifican por la ubicación del gen (número). Por eso, al referirse al CYP1A2, se entiende que es la isoforma de la familia 1 y subfamilia A que se codifica en el segundo gen (Court, 2013; Lemke et al., 2013). A pesar de que la mayor parte de la información que se tiene sobre los sistemas metabólicos se encuentran en la literatura humana, el entendimiento de los sistemas de biotransformación en perros ha crecido y hoy se sabe que existen diferencias de importancia entre humanos y perros a este nivel. Como se puede observar en el Cuadro 4, existe una múltiple cantidad de isoformas de CYP identificadas en perros, algunas de las cuales presentan SNPs, pero de las que se tiene poco conocimiento de sus implicaciones en la clínica.
Cuadro 4 Isoformas de CYP caninos: ubicación, sustratos comunes y polimorfismos identificados
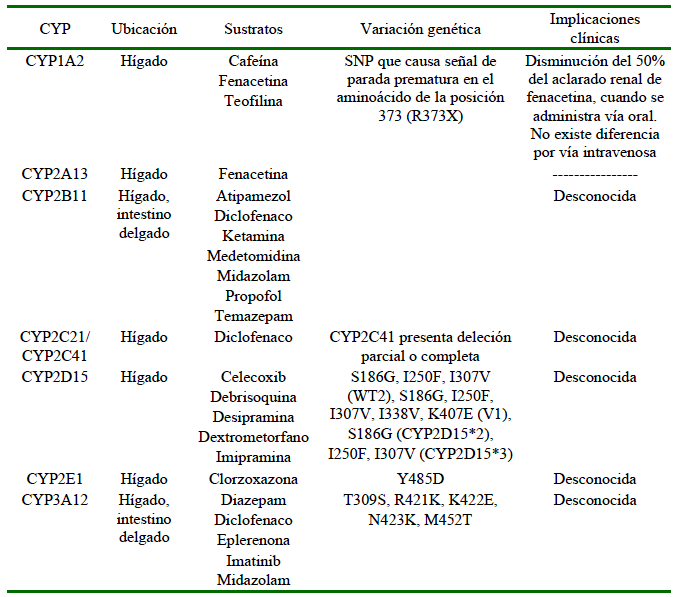
Elaboración propia, basado en datos de: Court, 2013; Martinez et al., 2020
En el caso del CYP2B11 se han dado reportes de variación en la actividad hasta 14 veces en perros de raza mixta (Mealey, 2006; Freishcher et al., 2008). La actividad del CYP2B11 en el Galgo es baja y esto permite que la concentración plasmática de propofol se mantenga constante por más tiempo en comparación a perros de raza mixta (Mealey, 2006). En los Beagle se han reportado posibles condiciones polimórficas, existiendo variaciones en el metabolismo de celecoxib, viéndose variaciones desde 1.5 a 5 h en el aclaramiento del fármaco, aunque esta variación no ha podido ser atribuida a ningún CYP especifico (Mealey, 2006).
El NADPH CYP450 oxidorreductasa (POR, por sus siglas en inglés), es un sistema de donación de electrones de todos los sistemas microsomales CYP, y del cual se han reportado dos variantes del gen no-sinónimas, que podrían influenciar una disminución en la actividad oxidasa del CYP2B11. Se reporta 36% de frecuencia de estas variantes en perros Lebrel Escocés y de 35% en el Galgo, en comparación a otras razas, y esto podría explicar la sensibilidad a anestésicos presente en la raza de perros Galgos (Mealey et al., 2019).
En referencia a las reacciones de fase II, se ha visto ausencia de genes codificantes para N-acetiltransferasas, en todas las razas de perros, lo que puede incrementar las reacciones de hipersensibilidad y efectos adversos asociados a sulfonamidas, procainamida, hidralazina y otros fármacos (Ruiz, 2001; Mealey, 2006). La enzima tiopurina metiltransferasa (TPMT, por sus siglas en inglés) que es responsable del metabolismo de fase II de drogas como la azatioprina presenta variaciones de actividad de hasta nueve veces, siendo los Schnauzer gigantes los que presentan la más baja actividad (Kidd et al., 2004). Una disminución en la actividad de la TPMT incrementa la susceptibilidad a la supresión de la médula ósea por azatioprina (Kidd et al., 2004; Mealey, 2006).
Polifarmacia e Interacciones Medicamentosas
El uso simultáneo de múltiples fármacos, la indicación de medicamentos de forma innecesaria, así como el uso de un fármaco para contrarrestar los efectos secundarios de otro, son parte de las consideraciones que se han acuñado alrededor del término polifarmacia en veterinaria (Homero, 2012; Hunter e Isaza, 2017). Al mezclar diferentes medicamentos es necesario que tanto el prescriptor como el farmacéutico conozcan los efectos esperados de cada fármaco, así como aquellos que podrían esperarse de la combinación de estos (Hunter e Isaza, 2017). En medicina veterinaria se carece de investigaciones referentes a esta temática; y existen pocos reportes sobre el uso simultáneo de fármacos en la práctica clínica de caninos. Según Hunter e Isaza (2017), la práctica de polifarmacia en medicina veterinaria es más común en pacientes crónicos, animales geriátricos con múltiples problemas y en protocolos anestésicos. En medicina humana se estima que cerca del 25% de los pacientes geriátricos se les prescribe fármacos de manera inapropiada (Zimmermann et al., 2013; Payne, 2016; Hunter e Isaza, 2017). La documentación de reacciones adversas a fármacos en veterinaria es más común que ocurra durante la fase de farmacovigilancia, debido a la carencia de estudios de gran escala, previo a la aprobación de un fármaco, como si ocurre en medicina humana (Hunter e Isaza, 2017). Según Alfaro-Mora (2018), 19.4% de los animales de compañía que va a consulta con médicos veterinarios en Costa Rica son polimedicados y, de estos, 21.6% presenta una potencial interacción medicamentosa.
Dentro de los fármacos que más destacan por su potencial inductor enzimático está el fenobarbital. Su administración oral o intravenosa puede alterar la farmacocinética de la fenitoína y la teofilina en perros Beagle, incrementándose su aclaramiento 2 y 3 veces, respectivamente (Sasaki y Shimoda, 2015). El fenobarbital también es capaz de inducir la enzima de fase II UDP-glucuroniltransferasa (UGT), en perros, y su administración conjunta con morfina incrementa hasta tres veces su glucuronidación hepática (Oguri et al., 1996). De la misma manera, antiinflamatorios no esteroideos (AINEs) como el carprofeno ven afectado su metabolismo, teniendo un aumento en su aclaramiento corporal (Sasaki y Shimoda, 2015). El omeprazol en perros es capaz de inducir los sistemas enzimáticos del CYP1A, mientras que la rifampicina lo hace sobre los CYP3A (Nishibe y Hirata, 1993; Sasaki y Shimoda, 2015).
Dentro de los medicamentos mejor caracterizados por su potencial inhibidor de los sistemas enzimáticos en perros está el ketoconazol y las fluoroquinolonas (Regmi et al., 2005; Aidasani et al., 2008). El CYP1A2 se ve inhibido por ciprofloxacino, orbifloxacino, enrofloxacino, ketoconazol, miconazol, fluvoxamina y ondansetron, el CYP2C21 es inhibido por vincristina, fluoxetina y clormipramina, el CYP2D15 es inhibido por la loperamida, vincristina, fluoxetina, ketoconazol y miconazol, y finalmente el CYP3A12 es inhibido por ketoconazol, miconazol, loperamida y ciclosporina (Regmi et al., 2005; Aidasani et al., 2008; Sasaki y Shimoda, 2015). Asimismo, se conoce que la dexametasona puede estar involucrada en un fenómeno de regulación descendente (down regulation), que puede causar interacciones droga-droga en sustratos metabolizados por el CYP2D y el CYP3A, y esto puede afectar los niveles plasmáticos de fármacos como la quinidina y el midazolam (Zhang et al., 2006a,b).
Panorama de la Medicina Individualizada
El primer test comercial de ADN para determinar la farmacogenética en perros estuvo disponible en 2004, y marcó lo que sería el inicio de la medicina individualizada en esta especie (Mealey et al., 2019). Sin embargo, la farmacogenética animal sigue rezagada con respecto a su contraparte humana. La falta de inversión en investigación y una disponibilidad relativamente reciente del genoma canino denotan este lento progreso en la comprensión real de las implicaciones de la farmacogenética en perros (Lindblad-Toh et al., 2005).
En medicina humana se habla de medicina de precisión, cuando se ha logrado caracterizar variaciones específicas en el genoma que afectan la respuesta a fármacos y a gracias a esto se establecen terapias específicas para el tratamiento de condiciones en las que es posible escoger una terapia determinada y que se sabe será efectiva frente a la variación genotípica de ese paciente (Kent et al., 2016). Un ejemplo de esto son las terapias contra algunos tipos de cáncer, donde la Administración de Drogas y Alimentos (FDA, por sus siglas en inglés) han aprobado drogas específicas para determinadas neoplasias (Tsimberidou et al., 2020). Un acercamiento a la medicina de precisión en perros puede referirse al uso de toceranib (inhibidor del receptor de tirosina quinasa), el cual está aprobado por la FDA para el tratamiento del tumor cutáneo de mastocitos en grado II y III, neoplasia común en perros. El toceranib funciona de una manera más eficiente en aquellos perros que presenten una mutación en el gen c-kit, reportándose una respuesta al tratamiento del 60% vs el 31% de aquellos que no la tienen (London, 2009, Klopfleisch, 2015).
A pesar de que el concepto de medicina personalizada no es algo novedoso, en la medicina veterinaria se requiere un mayor estudio, ya que aún es un campo incipiente y presenta muchos vacíos de conocimiento en la práctica y únicamente a través de un mayor esfuerzo investigativo sobre las variables genómicas que afectan el comportamiento de los fármacos en las diferentes razas caninas se puede lograr una aplicación real está en la práctica clínica.

