










Services on Demand
Journal

Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO  uBio
uBio
Share

 Permalink
PermalinkRevista Peruana de Biología
On-line version ISSN 1727-9933
Rev. peru biol. vol.25 no.3 Lima July/Set. 2018
http://dx.doi.org/10.15381/rpb.v25i3.15208
TRABAJOS ORIGINALES
Determinación de secuencia y modelaje por homología de la lipasa de Marinobacter sp. LB
Sequence determination and homology modeling of lipase from Marinobacter sp. LB
Jhosep Avila1, Amparo Iris Zavaleta1, Mercedes Palomino1, Christian Solis-Calero2
1 Universidad Nacional Mayor de San Marcos, Facultad de Farmacia y Bioquímica, Lab. de Biología Molecular. Jirón Puno 1002 Cercado de Lima, Perú.
2 Departamento de Biologia Estrutural e Funcional. Universidade Estadual de Campinas.
Resumen
Las lipasas de la familia I son reconocidas a nivel industrial por sus actividades catalíticas de esterificación, interesterificación y transesterificación. En esta investigación se caracterizó por análisis in silico a la lipasa de Marinobacter sp. LB aislado de las Salinas de Pilluana, San Martín. Con tal finalidad, se amplificó el gen lip mediante la reacción en cadena de la polimerasa (PCR) de punto final y la secuencia nucleotídica fue analizada in silico. Se elucidó la estructura terciaria empleando como molde a la lipasa 1EX9 de Pseudomonas aeruginosa PAO1 y se ejecutó el acoplamiento molecular con tres sustratos. El gen lip presentó 927 pb y la proteína madura, 284 aminoácidos. La lipasa posee un peso molecular de 29.99 kDa y un pI de 8.89. Asimismo, se identificaron residuos Ser78, Asp229 e His251, típicos de la triada catalítica de una lipasa de la familia I. Además, se evidenciaron once α-hélices periféricas y siete láminasβ internas. La región del bolsillo de unión y su afinidad por lípidos fue demostrada realizando acoplamientos moleculares con trioctanoina, tributirina y trioleina, con energías de -314.28, -248.11 y -215.44 kcal/mol, respectivamente; siendo los aminoácidos de interacción Asn167, Lys106, Trp172, Thr164, Ala179. En conclusión, la estructura tridimensional de la lipasa de Marinobacter sp. LB fue construida por modelamiento homólogo y validada en base a la calidad estereoquímica y el entorno de sus aminoácidos; mientras que, los análisis de acoplamiento con sustratos de lipasas permitieron evidenciar los aminoácidos que participan en el bolsillo de unión.
Palabras clave: Lipasa; Marinobacter; análisis in silico.
Abstract
Family I lipases are industrially recognized for their catalytic activities of esterification, interesterification and transesterification. In this study, Marinobacter sp. LB lipase isolated from Salinas de Pilluana, San Martín was characterized by in silico analysis. For this purpose, lip gene was amplified by conventional Polymerase Chain Reaction (PCR) and nucleotide sequence was analyzed in silico. The tertiary structure was elucidated using the 1EX9 lipase from Pseudomonas aeruginosa PAO1 as a template and molecular docking was executed with three substrates. The lip gene had 927 bp and mature protein, 284 amino acids. The lipase had a molecular weight of 29.99 kDa and pI of 8.89. Also typicall catalytic triad residues of family I lipases (Ser78, Asp229 and His251) were identified. In addition, eleven peripheral α-helixs and seven internal β-sheets were found. Binding pocket and its affinity for lipids were demonstrated by making molecular couplings with trioctanoin, tributyrin and triolein, with energies of -314.28, -248.11 and -215.44 kcal/mol, respectively; amino acids of interaction being Asn167, Lys106, Trp172, Thr164, Ala179. In conclusion, a 3D structure of Marinobacter sp. LB lipase was built using homologous modeling and validated based on the stereochemical quality and amino acids environment; while docking analysis with lipases substrates allowed to demonstrate the amino acids that participate in the binding pocket.
Keywords: Lipase; Marinobacter; in silico analysis.
Introducción
Las lipasas (EC 3.1.1.3) son un grupo de enzimas con gran interés industrial, científico y tecnológico, debido a su capacidad catalítica en reacciones de esterificación, interesterificación y transesterificación en medios no acuosos (Houde et al. 2004). En ese sentido, son utilizadas en las industrias: farmacéutica, cosmética, médica, alimentaria, oleoquímica, química, papelera, textilera, entre otras (Louwrier 1998, Schiraldi et al. 2002, Hasan et al. 2006, Wang et al. 2008, Monsia & Monsia 2013).
La primera clasificación de lipasas bacterianas realizada por Jaeger et al. (1999) consideró seis familias, dentro de las cuales destaca la familia I, verdaderas lipasas, con su miembro más representativo, la de Pseudomonas aeruginosa. En los meses sucesivos, la clasificación se extendió con las familias VII y VIII (Arpigny & Jaeger 1999).
Las características de las verdaderas lipasas según Kim et al. (2004) son:
-
La triada catalítica conformada por serina, ácido aspártico e histidina, donde la serina está dentro del pentapéptido conservado Gly-X-Ser-X-Gly.
-
La región del agujero oxianión constituida por los residuos His-Gly y ubicada en la región amino terminal, cadena arriba entre 70 a 100 aminoácidos (aa) del pentapéptido.
-
La secuencia señal localizada entre 10 a 40 aminoácidos por encima de la secuencia His-Gly.
Las propiedades y características de las lipasas se pueden determinar por diversas metodologías, entre las más utilizadas son las técnicas moleculares que permiten identificar, caracterizar y expresar genes específicos en hospederos conocidos y fáciles de manipular (Jorgensen et al. 1991, Ihara et al. 1991, Tan & Miller 1992, Saeed et al. 2006, Ruiz et al. 2002, An et al. 2003).
Por otro lado, los análisis in silico permiten predecir de manera rápida y económica algunas propiedades de las enzimas, tales como el peso molecular, punto isoeléctrico (pI), estructura tridimensional, sitio activo, especificidad de sustratos, entre otros. Así, Wohlfarth et al. (1992) clonaron el gen lipA de Pseudomonas aeruginosa PAO1 de 936 pares de bases (bp) determinando con métodos bioinformáticos un peso molecular de 30.134 kDa y la presencia del pentapéptido Gly-His-Ser-His-Gly. De forma similar, Alquati et al. (2002) amplificaron y clonaron el gen de una lipasa de 879 bp perteneciente a Pseudomonas fragi IFO3458 y mediante análisis in silico determinaron una secuencia proteica de 293 aa, con 32.086 kDa y pI de 9.33. Adicionalmente, Messaoudi et al. (2011) obtuvieron de la base de datos NCBI una lipasa de Arabidopsis thaliana de 379 aa y 44.23 kDa con la finalidad de generar un modelo de homología, para lo cual eligieron como molde una lipasa humana con 32% de identidad. La estructura fue validada y sometida al análisis de acoplamiento molecular identificando dos bolsillos de interacción con los sustratos ensayados. Posteriormente, Ali et al. (2013) modelaron una lipasa de Pseudomonas sp. AMS8 de 476 aa y 50 kDa, extraída de la base de datos, en este estudio, utilizaron como posibles plantillas a las lipasas de Serratia marcescens (80% de identidad) y Pseudomonas sp. MIS38 (69% de identidad); sin embargo, sus análisis generaron un mejor modelo con la lipasa de Pseudomonas sp. MIS38, evidenciando que los porcentajes de identidad altos no brindan necesariamente los mejores modelos. Por último, Gupta et al. (2015) y Lanka et al. (2015), realizaron el modelamiento homologo, la validación de la estructura y los estudios de acoplamiento de lipasas de Pseudomonas fluorescens y Emericella nidulans NFCCI 3643, respectivamente. En ambos casos, el bolsillo de interacción fue único para los sustratos ensayados.
Marinobacter sp. LB es una bacteria halófila moderada, aislada de las Salinas de Pilluana (San Martín-Perú), presenta una lipasa extracelular versátil ante condiciones de estrés salino (FernándezJerí et al. 2013). Sin embargo, el cultivo de esta bacteria en medios salinos dificulta los procesos de filtración, precipitación, diálisis y cromatografía, evitando su caracterización total. Por este motivo se decidió realizar el análisis in silico del gen lip y de la secuencia aminoacídica, con la finalidad de pronosticar la estructura de la enzima, sus características y su mecanismo de interacción con sustratos.
Material y métodos
Extracción de ADN genómico. Se utilizó Marinobacter sp. LB proveniente del cepario del Laboratorio de Biología Molecular de la Facultad de Farmacia y Bioquímica de la Universidad Nacional Mayor de San Marcos. A partir de la cual se prepararon cultivos frescos de 12 h a 37 °C en caldo tripticasa de soya y NaCl al 3%. A continuación, se centrifugó 1.5 mL de cultivo a 12000 rpm por 10 min, eliminando el sobrenadante. El pellet fue resuspendido con 200 µL de tampón PBS 1X y NaCl 3%, se centrifugó nuevamente a 12000 rpm por 10 min, eliminando el sobrenadante. Con la finalidad de lisar las membranas celulares, se adicionaron 200 µL de agua bidestilada y las células fueron sometidas a un choque térmico de 100 °C por 10 min y -20 °C por 2 min. Luego, se centrifugó a 12000 rpm por 10 min y se conservó el sobrenadante. Para verificar la calidad del ADN genómico, se realizó una electroforesis en gel de agarosa 1% (Biorad) con tampón TAE 1X y marcador DNA Ladder 1 kb (Invitrogen).
Amplificación del gen lip mediante PCR. Para la amplificación del gen lip mediante PCR se utilizaron los cebadores forward 5’-ATACTCCCTGTTGCATGTCG-3’ y reverse 5’-TGGACTGGTGCTTCATAACC-3’ diseñados con el programa primer3 (Untergasser et al. 2012, Koressaar & Remm 2007). El volumen final fue de 25 µL y la mezcla de reacción contenía KCl 50 mM, Tris/HCl 10 mM, tritón X-100 0.1% (v/v), MgCI2 1.5 mM, dNTPs 200 µM, 20 ρmoles de cada cebador, Taq ADN-polimerasa 1 U y 50 ng de ADN genómico. La PCR se inició con una desnaturalización del ADN a 94 °C por 4 min seguido de 35 ciclos repetitivos a 94 °C por 45 s, 45 °C por 55 s y 72 °C por 45 s, con una extensión final a 72 °C por 7 min. La verificación de los productos amplificados se realizó por electroforesis en gel de agarosa 1% con tampón TBE 0.5X. La secuencia nucleotídica del gen lip de Marinobacter sp. LB se obtuvo con el método de Sanger a través de los servicios de secuenciación de la Universidad Peruana Cayetano Heredia.
Análisis del gen lip y modelado de la Lipasa de Marinobacter sp. LB.- La transcripción y traducción del gen lip, se realizó con el programa Bioedit (Hall 1999) considerando un marco de lectura +1. La secuencia de aminoácidos fue sometida a un alineamiento local utilizando la herramienta BLAST, de la base de datos GenBank, y se extrajo algunas secuencias de lipasas de la Familia I.
Posteriormente, se realizó un alineamiento múltiple con el programa ClustalX 2.1 (Thompson et al. 1997). Se compararon 21 secuencias de lipasas, entre las cuales figuraba la secuencia de Marinobacter sp. LB, las del GenBank y lipasas representativas de la Familia I obtenidas de Arpigny y Jaeger (1999). Este segundo alineamiento se realizó con la finalidad de verificar la familia a la que pertenece nuestra enzima y elucidar las regiones conservadas como el pentapéptido Gly-X-Ser-X-Gly.
Con ayuda del programa SignalP (Petersen et al. 2011), se determinó el péptido señal en la región amino terminal de la proteína en estudio. Para éste análisis se consideró que Marinobacter sp. LB es una bacteria Gram negativa y la lipasa producida es extracelular (Chávez-Hidalgo 2010, Fernandez-Jerí et al. 2013).
Selección de la estructura molde.- Para seleccionar a la mejor estructura molde, se buscaron secuencias proteicas obtenidas a partir de estudios cristalográficos en las bases de datos Protein Data Bank (PDB) y UniProtKB. Las secuencias encontradas se compararon con la obtenida en este estudio. El molde elegido fue la lipasa codificada como 1EX9 (secuencia plantilla), perteneciente a Pseudomonas aeruginosa PAO1.
Modelamiento homólogo.- El modelamiento homólogo se construyó utilizando el programa Swiss-PDB Viewer 4.0.4 (Guex & Peitsch 1997) en modo proyecto, el cual permite analizar la estructura tridimensional de las proteínas. Este análisis se basa en el alineamiento entre la secuencia blanco (secuencia en estudio) con la plantilla. Durante este proceso, se identifican las estructuras secundarias y terciarias de la plantilla, mapeando los residuos similares con la secuencia blanco. Así, se alinearon las secuencias, luego se realizó la inspección visual y se editó manualmente el alineamiento, para aumentar sitios conservados y finalmente, se superpuso las secuencias de aminoácidos. Asimismo, se calculó la raíz de la desviación cuadrática media (RMSD) con el mismo programa para evaluar la confiabilidad de la estructura modelada.
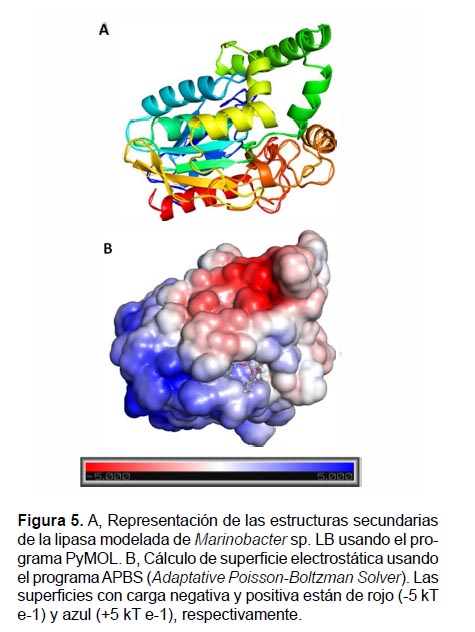
La superficie electrostática de la lipasa modelada se determinó empleando APBS (Adaptive Poisson-Boltzmann Solver) que realiza cálculos electrostáticos de Poisson-Boltzmann sobre biomoléculas (Baker et al. 2001) para describir la complementariedad ligando-proteína basado en la estructura geométrica e interacciones electrostáticas pues desempeñan un papel importante en la catálisis enzimática. El modelo se visualizó utilizando el programa PyMOL (DeLano 2002).
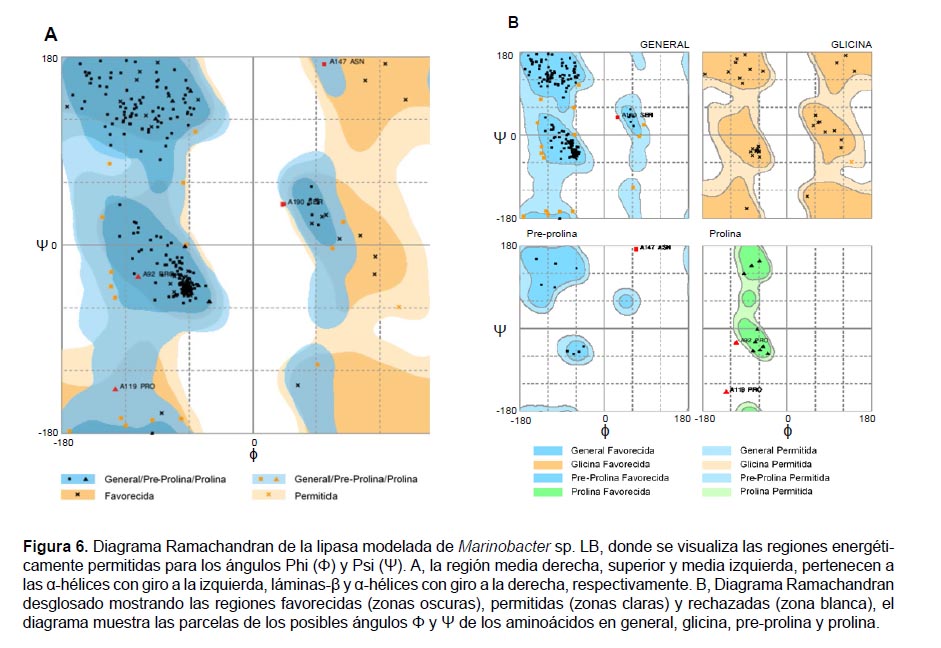
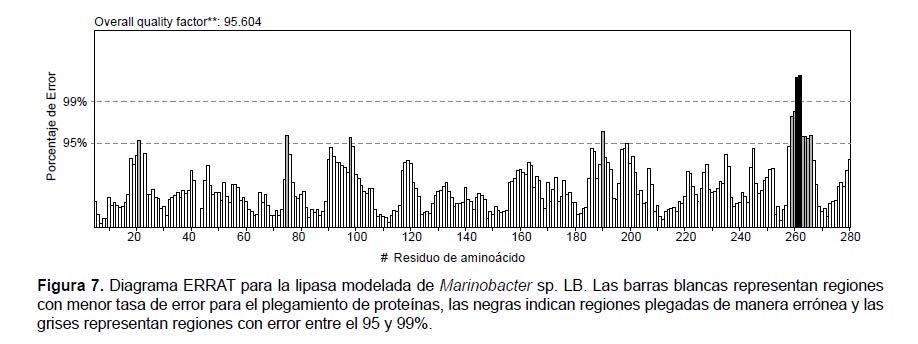
Validación del modelamiento.- El proceso de validación se realizó para verificar que el modelado de la lipasa cumple con los requisitos mínimos de calidad. En consecuencia, el modelo fue sometido a los análisis de: evaluación de la calidad estereoquímica de los puntos de Ramachandran, con el programa RAMPAGE (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php); evaluación del entorno de los aminoácidos considerando la calidad de la estructura proteica, con Verify 3D (Bowie et al. 1991, Lüthy et al. 1992) y análisis de las regiones plegadas, utilizando ERRAT (Colovos & Yeates 1993). Los dos últimos programas estuvieron en el servidor del Instituto DOE de la Universidad de California, Los Ángeles (http://www.doe-mbi.ucla.edu/).
Acoplamiento molecular.- Con la finalidad de obtener las energías de acoplamiento y el posible modo de interacción entre la lipasa de Marinobacter sp. LB y los tres sustratos seleccionados, se realizaron los dos análisis descritos a continuación:
El primero se realizó con el programa HEX 8.0.0 (Ritchie et al. 2008), los sustratos fueron tributirina (CID: 6050), trioctanoina (CID: 10850) y trioleina (CID: 11970724), extraídos de la base de datos de moléculas químicas del NCBI (PubChem). Los parámetros utilizados fueron predeterminados por el programa que simuló varias orientaciones posibles con sus respectivas energías de acoplamiento, brindando el modelo más viable. El tipo de correlación y la salida del post procesamiento para el receptor y el ligando se mantuvieron con base a la forma, el potencial electrostático y la minimización de la mecánica molecular (MM). El acoplamiento se llevó a cabo a rotación total del ligando manteniendo la posición del receptor fija en el espacio. Este programa fue validado reproduciendo tres modelos cristalográficos de lipasas pertenecientes a Pseudomonas aeruginosa (PDBID: 1EX9), Burkholderia cepacia (PDBID: 1YS1) y Pseudomonas cepacia (PBDID: 1HQD) en interacción con RC(RP,SP)-1,2-dioctil carbamoil-glicero-3-O-octilfosfonato (CID: 445454), ácido hexilfosfónico (R)-2-metil-3-fenil propil ester (CID: 4369425) y 1-fenoxi-2-acetoxibutano (CID: 446123), respectivamente. Los acoplamientos para la validación fueron realizados retirando los sustratos de los modelos cristalográficos con ayuda del programa Swiss-PDB Viewer 4.0.4 (Guex & Peitsch 1997) y acoplándolos nuevamente con el programa HEX 8.0.0 (Ritchie et al. 2008). Además, las interacciones se analizaron con el programa Discovery Studio 2017 R2 (Dassault Systèmes BIOVIA, San Diego, CA, USA).
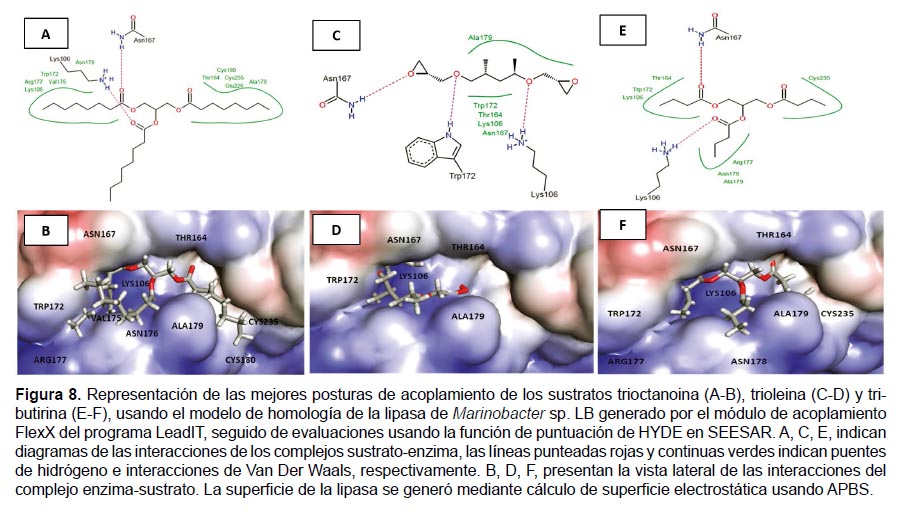
El segundo análisis de acoplamiento para tributirina, trioctanoina y trioleina fue realizado con el módulo de acoplamiento FlexX del programa LeadIT 2.1.8 (https://www.biosolveit.de/); los resultados fueron evaluados usando la función de puntuación HYDE en SEESAR 70 (https://www.biosolveit.de/). El punto de partida fue la estructura obtenida por modelamiento homólogo de la lipasa de Marinobacter sp. LB. Para éste estudio, el sitio de unión en la proteína se limitó a 10 Å y las 50 soluciones con mejores puntuaciones (FlexX) de cada ligando fueron conservadas y posteriormente analizadas para obtener las puntuaciones subsecuentes con SEESAR; por último, se seleccionaron las posturas con las mejores puntuaciones.
Resultados
Amplificación del gen lip. A partir del ADN amplificado y separado en gel de agarosa 1% (Fig. 1), se calculó que el gen lip de Marinobacter sp. LB midió aproximadamente 998 pb. Este cálculo fue realizado por regresión lineal y el producto amplificado fue secuenciado. El electroferograma mostró picos bien definidos para cada base, sin ruido ni superposiciones.
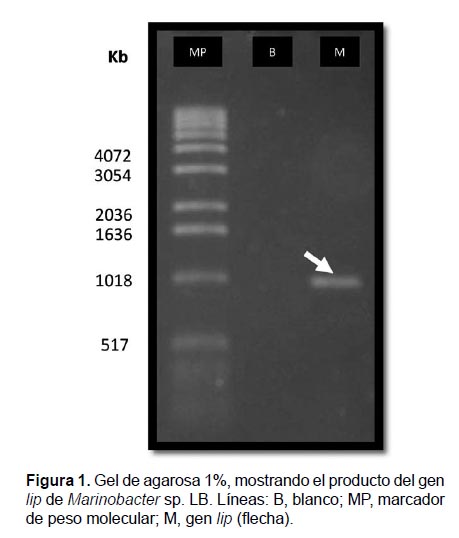
Análisis del gen lip y modelamiento de la lipasa de Marinobacter sp. LB.La secuencia génica de la lipasa de Marinobacter sp. LB, desde el codón de inicio hasta el de término, fue de 927 nucleótidos y se analizó con Bioedit (Hall 1999) para obtener una cadena aminoacídica de 308 aminoácidos (material complementario 1).
Para el estudio de la estructura primaria se utilizaron 21 secuencias proteicas: once descritas por Arpigny y Jaeger (1999); nueve obtenidas de la base de datos GenBank y una perteneciente a nuestra lipasa. De todas las enzimas, las secuencias de Pseudomonas aeruginosa ATCC31156, Burkholderia glumaeCBS322.89, Burkholderia cepacia, Chromobacterium viscosumATCC6918 son representativas de la familia I (Tabla 1).
El tamaño de la proteína inmadura es de 308 aa, constituida en mayor proporción por glicina (11%), alanina (9.4%) y leucina (9.1%); en forma minoritaria, por ácido glutámico (1.9%), triptófano (1.9%) y cisteína (0.6%). La secuencia del péptido señal detectada por SignalP (Petersen et al. 2011) está en la región N-terminal de la proteína, compuesta por los primeros 24 aa. Por lo tanto, la lipasa madura extracelular tendría un tamaño teórico de 284 residuos con peso molecular de 29.99 kDa, pI de 8.89. En base al alineamiento múltiple, se pronosticó que los aminoácidos catalíticos de nuestra lipasa son Ser78, Asp229 e His251. Mientras que, la Cys180 y Cys235 participan en la formación del puente disulfuro. Cabe señalar que las lipasas son dependientes de calcio (Jaeger et al. 1999), siendo los posibles residuos de unión a este cofactor Asp210 y Asp253 (Fig. 3).

Para la selección de la estructura molde y posterior modelamiento de la lipasa se comparó la secuencia aminoacídica en estudio con las bases de datos (PDB, UniProtKB) y se encontró seis proteínas equivalentes provenientes de diferentes especies bacterianas (Tabla 2). De las cuales, Pseudomonas aeruginosa PAO1 (43%), Proteus mirabilis (37%) y Pseudomonas glumae (34%) presentan estudios por cristalografía. Los análisis evolutivos demuestran que las especies de Marinobacter tienen mayor relación evolutiva con Pseudomonas aeruginosa y menor relación con el género Proteus (Anzai et al. 2000). El porcentaje de identidad, la estructura cristalográfica y la relación evolutiva fueron los factores que determinaron a la lipasa 1EX9, perteneciente a Pseudomonas aeruginosa PAO1, como molde (plantilla) para el modelaje por homología.
El modelo fue construido con el programa Swiss-PDB Viewer (Guex & Peitsch 1997), para lo cual, primero se realizó el alineamiento de nuestra secuencia madura con la lipasa 1EX9 de Pseudomonas aeruginosa PAO1. Durante la ejecución del programa, se logró reconocer las estructuras secundarias propias de la plantilla, en base a su estructura cristalográfica. Por tal motivo, en las regiones donde los aminoácidos de ambas secuencias coincidían, se construyó la estructura terciaria de la lipasa de Marinobacter sp. LB. La raíz de la desviación cuadrática media (RMSD) obtenido con el mismo programa, en donde se ajustaron los carbonos alfa de la estructura en mención, fue 0.32 Å.
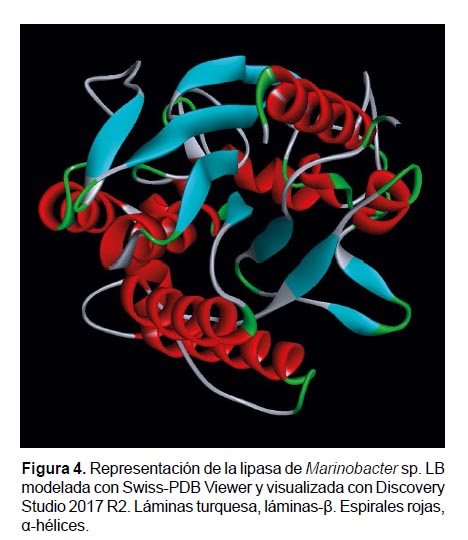
La representación gráfica del modelaje por homología de la lipasa se observó con el programa Discovery Studio 2017 R2 (Dassault Systèmes BIOVIA, San Diego, CA, USA), diferenciando que 11 α-hélices se encuentran distribuidas en toda la proteína, mientras que 7 de las 9 láminasβ se agrupan en la región central de la biomolécula (Fig. 4). La lipasa madura de Marinobacter sp. LB no presenta el péptido señal, motivo por el cual el porcentaje de identidad con la lipasa (PBDID: 1EX9) aumenta a 45.85%. En la Figura 4 se observa que nuestra enzima es monomérica. La superficie electrostática de la enzima (Fig. 5) evidencia que la polaridad del ligando y de la superficie de la lipasa modelada, son complementarias.
Validación del modelamiento. Previo al acoplamiento molecular, el modelo desarrollado fue validado mediante la evaluación de la calidad estereoquímica y del entorno de los aminoácidos. En la primera evaluación, el programa RAMPAGE generó el diagrama de Ramachandran (Fig. 6) a partir de la conformación de los aminoácidos, en base a los giros que existen entre los enlaces del carbono alfa con los grupos amino y carboxilo, denominados ángulos phi (Φ) y psi (Ψ), respectivamente. La figura 6A presenta tres regiones definidas, evidenciando la presencia de láminasβ, α-hélices con giros a la derecha e izquierda. La figura 6B indica la posición de los aminoácidos, en donde el 92.6% están en la región "favorecida" (zonas oscuras), 6% en la "permitida" (zonas claras) y 1.4% en la "desfavorable" (zonas blancas).
En la segunda evaluación, el programa ERRAT (Colovos & Yeates 1993) identificó la presencia de regiones incorrectamente plegadas, pues 12 aa pertenecen a regiones inconsistentes. Sin embargo, 272 aa forman parte de regiones plegadas dentro del límite permitido (Fig. 7), por lo que el programa brindó un "Factor de calidad general" del 95.604 %. Por otra parte, se analizó la compatibilidad de la estructura terciaria (3D) con la primaria (1D) usando el programa Verify 3D (Bowie et al. 1991, Lüthy et al. 1992), con puntuaciones que varían desde -1 (puntaje malo) hasta +1 (puntaje bueno), en este caso la compatibilidad fue del 78.52% que representa un "puntaje promedio 3D-1D" ≥ +0.2.
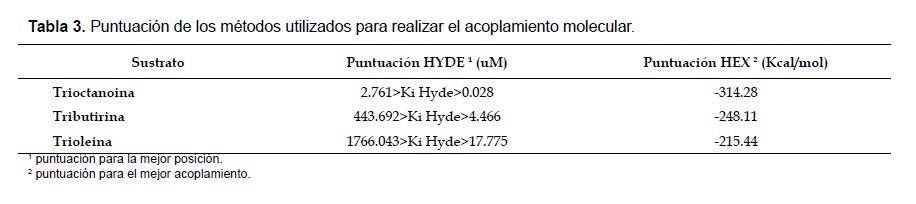
Acoplamiento molecular.Las energías de acoplamiento molecular fueron determinadas enfrentando la lipasa modelada con los sustratos. Para este fin, se realizó evaluaciones con el programa HEX (Ritchie et al. 2008) en donde se emplearon 2000 soluciones de acoplamiento en cada caso y se obtuvo las energías -248.11, -314.28 y -215.44 kcal/mol para tributirina, trioctanoina y trioleina, respectivamente (Tabla 3).
Las mismas condiciones fueron utilizadas para validar el programa HEX (Ritchie et al. 2008), donde se reprodujo el acoplamiento de las lipasas 1EX9, 1YS1 y 1HQD con sus respectivos sustratos, dando energías de acoplamiento muy bajas, lo que significa que existe una alta probabilidad para estas interacciones. Con el uso del programa Discovery Studio 2017 R2, se encontró que varios aminoácidos de cada bolsillo de unión de las estructuras cristalográficas (Tabla 4) también participaban del reconocimiento de los ligandos en los modelos pronosticados.

Además, el programa HEX (Ritchie et al. 2008) brindó coordenadas de interacción enzima-sustrato para la lipasa de Marinobacter sp. LB obteniendose valores muy similares para tributirina, trioctanoina y trioleina. Éstas coordenadas en conjunto con el modelo homólogo de la lipasa fueron utilizadas como punto de partida en los estudios de acoplamiento molecular realizados con el programa con LeadIT, el cual reveló que los tres sustratos se unen en el mismo bolsillo de unión (Fig. 8). La postura de los sustratos en la región de la enzima fue evaluada usando el programa HYDE, donde las puntuaciones son producto de la predicción de la afinidad de los ligandos a la proteína mediante los valores de la constante de disociación Ki (Tabla 3), un menor valor de Ki representa una mayor afinidad de la enzima por su sustrato, en consecuencia esto significa que las bajas concentraciones de sustrato pueden ser reconocidas por la enzima y ejercer su efecto. Los aminoácidos del bolsillo que interaccionan con los tres sustratos son Asn167, Lys106, Trp172, Thr164, Ala179, además Arg177 y Cys235 coinciden para tributirina y trioctanoina, tal como se muestra en la Figura 8.
Discusión
La clasificación de las lipasas fue propuesta por Arpigny y Jaeger (1999) resaltando regiones conservadas en la familia I las que contienen a los aminoácidos de la triada catalítica, las mismas que están en la lipasa en estudio (Fig. 2). Por tal motivo, se podría considerar a esta enzima dentro del grupo de las verdaderas lipasas. La secuencia aminoacídica inmadura es de 308 residuos, el tamaño de estas proteínas depende de la familia a la que pertenece, la especie y del espacio donde actúa. Así mismo, las lipasas extracelulares bacterianas por lo general presentan un péptido señal (Ihara et al. 1991, Frenken et al. 1992, Sangeetha et al. 2014, Sullivan et al. 1999, Park et al. 2009) en la región N-terminal, reconocido por receptores de membrana y proteínas de secreción que permiten su salida al medio externo (Jaeger et al. 1999). En tal sentido, el reconocimiento de un péptido señal de 24 aa en la secuencia de la lipasa, se puede sustentar con los trabajos realizados por Alejandro-Paredes (2012) y FernándezJerí et al. (2013) quienes describen que la lipasa de Marinobacter sp. LB es extracelular.

La secuencia madura de la lipasa presentó 284 residuos, 29.99 kDa y pI de 8.89. Los aminoácidos de la triada catalítica son Ser78, Asp229 e His251, también se reconocieron los residuos de unión al Ca2+ en las posiciones Asp210 y Asp253. Hallazgos similares fueron reportados por Ihara et al. (1991) quienes describen lipasas de Pseudomonas nov. sp. 109, cuya secuencia madura presenta 311 aa, 30.149 kDa y pI de 7; Frenken et al. (1992) caracterizaron una lipasa de Pseudomonas glumae PG1, con una secuencia madura de 319 aa, 33.1 kDa y pI de 7.2, los residuos catalíticos descritos fueron Ser87, Asp241 e His285; Sullivan et al. (1999) presentaron una lipasa de Acinetobacter calcoaceticus RAG-1 de 313 aa, 32.5 kDa y pI de 6.16, identificaron los residuos Asp240 y Asp281 como los de unión al Ca2+, mientras que los aminoácidos catalíticos fueron Ser111, Asp258 e His280; Alquati et al. (2002) reportaron una lipasa de Pseudomonas fragi IFO 3458 con 293 aa, 32.086 kDa y pI de 9.33, la triada catalítica fue Ser83, Asp238 e His260; An et al. (2003) identificaron una lipasa de Pseudomonas sp. SW-3, con 311 aa y 31 kDa, los residuos catalíticos fueron Ser108, Asp255 e His277, y los residuos de unión al Ca2+ descritos como Asp240 y Asp279.
El porcentaje de identidad entre la lipasa en estudio con la de Pseudomonas aeruginosa PAO1 fue de 45.85%, otros modelos como el desarrollado por Gupta et al. (2015) utilizan 80.5% de identidad entre su lipasa y la estructura molde. Sin embargo, un porcentaje mayor no asegura el desarrollo de un buen modelo homólogo (Ali et al. 2013). Así lo demuestran Messaoudi et al. (2011) y Lanka et al. (2015) quienes modelaron lipasas con identidades del 32% y 32.77%, respectivamente. Más aún, Yang et al. (2000) demuestran que porcentajes de identidad mayores al 25% indican estructuras tridimensionales semejantes entre dos proteínas.
La estructura tridimensional de la lipasa de Marinobacter sp. LB, presentó 11 α-hélices periféricas y 9 láminasβ ubicadas principalmente en la región central, similar a lo descrito por Messaoudi et al. (2011) quienes describen en su modelo 8 láminasβ centrales. El plegamiento y estabilidad de nuestra lipasa depende de: los puentes de hidrógeno, entre aminoácidos polares; interacciones hidrofóbicas, entre residuos apolares (glicina, alanina, valina, leucina, isoleucina, prolina); y un puente disulfuro generado entre los grupos tiol (-SH) de los residuos Cys180 y Cys235, también descrito por Frenken et al. (1992), An et al. (2003), Sangeetha et al. (2014) y Sullivan et al. (1999). En relación al RMSD, los modelos que presentan un porcentaje de identidad entre 90% a 95%, suelen tener valores entre 1 y 0.5 Å (Rost 1999, Vivas-Reyes et al. 2010), por lo que puntuaciones inferiores a 0.5 Å deben considerarse estructuralmente confiables, tal como sucede con el valor de 0.32 Å obtenido con Swiss-PDB Viewer (Guex & Peitsch 1997).
En la validación de la estructura de la lipasa (Fig. 4), el programa RAMPAGE determinó tres regiones: favorecida (92.6%), permitida (6%) y atípica (1.4%). Cabe resaltar que el programa recomienda valores aproximados al 98% en la región favorecida, sin embargo, el puntaje obtenido es aceptable por ser un modelo predictivo y no una estructura cristalográfica completamente resuelta (Ali et al. 2013). Al respecto, Messaoudi et al. (2011) y Ali et al. (2013), mencionan que valores superiores al 90% indican modelos con buena calidad estereoquímica.
El programa ERRAT (Colovos & Yeates 1993) asignó un "Factor de calidad general" de 95.60% que indica un buen modelo estructural debido a que el programa recomienda valores superiores a 95%. A pesar de esto, se han validado estructuras con 84.67% (Lanka et al., 2015). Por otro lado, el programa Verify 3D muestra que el 78.52% de los aminoácidos presenta un "Puntaje promedio 3D-1D" mayor a +0.2. En este contexto, Bowie et al. (1991) y Lüthy et al. (1992), indican que por lo menos el 65% de aminoácidos deben tener puntajes mayores a +0.2, lo cual demuestra una buena relación entre el modelo tridimensional y la estructura primaria. Así, todos los análisis reflejan un modelo predictivo adecuado de la lipasa de Marinobacter sp. LB.
En los análisis de acoplamiento, los tres sustratos se unieron al mismo bolsillo, en interacción con los aminoácidos Asn167, Lys106, Trp172, Thr164, Ala179, y parcialmente con Arg177, Cys235, Val175, Asn176, Cys180, Asn178. La ausencia de los residuos de la triada catalítica indica que los aminoácidos catalíticos son los más reactivos y en consecuencia importantes en la catálisis enzimática, pero no necesariamente intervienen en la unión enzima-sustrato. De manera similar, Gupta et al. (2015) describen el acoplamiento de una lipasa de Pseudomonas fluorescens con p-nitrofenil, ión acetato y dietil fosfonato, mediante los residuos Leu26, Tyr29, Asn31, Asp33, Pro315 y Thr316. Por otro lado, las energías de acoplamiento y constantes de disociación Ki (tabla 3) indican que la lipasa de Marinobacter sp. LB tiene mayor afinidad por la trioctanoina, seguido por tributirina y trioleina, puesto que la menor energía utilizada en la unión enzima-sustrato genera mayor probabilidad de acoplamiento (Ritchie et al. 2008) y valores bajos de la contante Ki indican que la enzima puede reconocer bajas concentraciones del sustrato y ejercer su efecto catalítico. Los estudios experimentales realizados por AlejandroParedes (2012), donde la lipasa de Marinobacter sp. LB degrada más tributirina que trioleina, refuerzan lo antes mencionado. Adicionalmente, describen que las lipasas interaccionan con bajas energías de acoplamiento con trioctanoina, tributirina y trioleina (Messaoudi et al. 2011, Lanka et al. 2015).
Conclusión
La lipasa de Marinobacter sp. LB fue construida por modelamiento homólogo, la estructura tridimensional fue validada en base a la calidad estereoquímica y el entorno de sus aminoácidos. Los análisis de acoplamiento con sustratos naturales de lipasas, permitieron evidenciar los aminoácidos que participan en el bolsillo de unión. Este trabajo propone alternativas para la caracterización parcial y total de proteínas a partir de secuencias génicas. Se recomienda para futuras investigaciones de esta lipasa, realizar estudios de optimización por dinámica molecular.
Agradecimientos
A BioSolveIT GmbH de la Universidad Estadual de Campinas por el acceso a los programas LeadIT 2.1.8 y Seesar 70 para el análisis quimioinformático.
Fuentes de financiamiento: Este estudio fue financiado parcialmente por los contratos 017-FINCYT-PIBAP-2008 y 007-FONDECYT-2014.
Información sobre los autores:
JA realizo las pruebas experimentales, análisis bioinformático. AIZ realizo la asesoría del trabajo, pruebas experimentales, análisis bioinformático. MP realizo las pruebas experimentales. CS-C realizo el análisis bioinformático. Todos los autores participaron en la redacción, revisión y aprobación del manuscrito.
Los autores no incurren en conflictos de intereses.
Literatura citada
Alejandro-Paredes A. 2012. Producción en cultivo discontinuo y caracterización parcial de lipasas de Marinobacter sp. aislada de las salinas de Pilluana [Tesis para obtener el título de Químico Farmacéutico]. Facultad de Farmacia y Bioquímica. Universidad Nacional Mayor de San Marcos. [ Links ]
Ali M.S.M., S.F.M. Fuzi, M. Ganasen, R.N.Z.R.A. Rahman, M. Basri & A.B. Salleh. 2013. Structural Adaptation of Cold-Active RTX Lipase from Pseudomonas sp. Strain AMS8 Revealed via Homology and Molecular Dynamics Simulation Approaches. BioMed Research International 2013: 1-9. https://doi.org/10.1155/2013/925373 [ Links ]
Alquati C., L. De Gioia, G. Santarossa, L. Alberghina, P. Fantucci & M. Lotti. 2002. The cold-active lipase of Pseudomonas fragi. Heterologous expression, biochemical characterization and molecular modeling. European Journal Biochemistry 269: 3321-3328. http://dx.doi.org/10.1046/j.14321033.2002.03012.x [ Links ]
An S.Y., S-W. Kim, Y.L. Choi, Y.S. Cho, W.H. Joo & Y.C. Lee. 2003. Cloning, expression in Escherichia coli, and enzymatic properties of a lipase from Pseudomonas sp. SW-3. Journal of Microbiology 41(2): 95-101. [ Links ]
Anzai Y., H. Kim, J-Y. Park, H. Wakabayashi, & H. Oyaizu. 2000. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. International Journal of Systematic and Evolutionary Microbiology. 50: 1563-1589. doi: 10.1099/00207713-50-4-1563 [ Links ]
Arpigny J.L. & K.E. Jaeger. 1999. Bacterial lipolytic enzymes: classification and properties. Biochemical Journal 343(1): 177-183. http://dx.doi.org/10.1042/bj3430177 [ Links ]
Baker N.A., D. Sept, S. Joseph, M.J. Holst & J.A. McCammon. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proceedings of the National Academy of Sciences of United States of America 98(18): 10037-10041. http://dx.doi.org/10.1073/pnas.181342398 [ Links ]
Bowie J.U., R. Lüthy & D. Eisenberg. 1991. A method to identify protein sequences that fold into a known three-dimensional structure. Science 253(5016): 164-170. http://dx.doi.org/10.1126/science.1853201 [ Links ]
Chávez-Hidalgo E. 2010. Bacterias halófilas moderadas con actividad lipolítica aisladas de las salinas de Pilluana-San Martín. [Tesis para obtener el grado de Magister]. Facultad de Farmacia y Bioquímica. Universidad Nacional Mayor de San Marcos. [ Links ]
Colovos C. & T.O. Yeates. 1993. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Science 2(9): 1511-1519. http://dx.doi.org/10.1002/pro.5560020916 [ Links ]
De Lano W.L. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA; http://www.pymol.org [ Links ]
Fernández-Jerí Y., A. Zavaleta, L. Alejandro-Paredes & V. Izaguirre. 2013. Caracterización parcial de una lipasa extracelular de Marinobacter sp. empleando la metodología de superficie respuesta. Ciencia e Investigación 16(1): 12-17. [ Links ]
Frenken L.G.J., M.R. Egmond, A.M. Batenburg, J.W. Bos, C. Visser & C.T. Verrips. 1992. Cloning of the Pseudomonas glumae lipase gene and determination of the active site residues. Applied Environmental Microbiology 58(12): 3787-3791. [ Links ]
Guex N. & M.C Peitsch. 1997. SWISS-MODEL and the SwissPdbViewer: an environment for comparative protein modeling. Electrophoresis 18(15): 2714-2723. http://dx.doi.org/10.1002/elps.1150181505 [ Links ]
Gupta G.N., V.K. Singh & V. Prakash. 2015. Molecular modeling and docking studies of cold active lipase from Pseudomonas fluorescens. International Journal of Applied Biology and Pharmaceutical Technology 6: 59-66. [ Links ]
Hall T.A. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series 41: 95-98. [ Links ]
Hasan F., A.A. Shah & A. Hameed. 2006. Industrial applications of microbial lipases. Enzyme and Microbial Technology 39: 235251. http://dx.doi.org/10.1016/j.enzmictec.2005.10.016 [ Links ]
Houde A., A. Kademi & D. Leblanc. 2004. Lipases and their industrial applications: an overview. Applied Biochemistry and Biotechnology 118(1-3): 155-170. http://dx.doi.org/10.1385/ABAB:118:1-3:155 [ Links ]
Ihara F., Y. Kageyama, M. Hirata, T. Nihira & Y. Yamada. 1991. Purification, characterization, and molecular cloning of lactonizing lipase from Pseudomonas Species. Journal of Biological Chemistry 266(27): 18135-18140. [ Links ]
Jaeger K.E., B.W. Dijkstra & M.T. Reetz. 1999. Bacterial biocatalysts: molecular biology, three dimensional structures, and biotechnological applications of lipases. Annual Review of Microbiology 53: 315-351. http://dx.doi.org/10.1146/annurev.micro.53.1.315 [ Links ]
Jorgensen S., K. Skov & B. Diderichsen. 1991. Cloning, sequence, and expression of a lipase gene from Pseudomonas cepacia: Lipase production in heterologous hosts requires two Pseudomonas genes. Journal of Bacteriology 173: 559-567. http://dx.doi.org/10.1128/jb.173.2.559-567.1991 [ Links ]
Kim H.K., Y-J. Jung, W-C. Choi, H.S. Ryu, T-K. Oh & J-K. Lee. 2004. Sequence-based approach to finding functional lipases from microbial genome databases. FEMS Microbiology Letters 235: 349-355. http://dx.doi.org/10.1111/j.1574-6968.2004.tb09609.x [ Links ]
Koressaar T. & M. Remm. 2007. Enhancements and modifications of primer design program Primer3. Bioinformatics 23(10): 1289-91. http://dx.doi.org/10.1093/bioinformatics/btm091 [ Links ]
Lanka S., V. R. Talluri, V. Ganesh & J.N.L. Latha. 2015. Homology Modeling, Molecular Dynamic Simulations and Docking Studies of a New Cold Active Extracellular Lipase, EnL A from Emericella nidulans NFCCI 3643. Trends in Bioinformatics 8(2): 37-51. http://dx.doi.org/10.3923/tb.2015.37.51 [ Links ]
Louwrier A. 1998. Industrial products: the return to carbohydratebased industries. Biotechnology and Applied Biochemistry 27: 1-8. http://dx.doi.org/10.1111/j.1470-8744.1998.tb01368.x [ Links ]
Lüthy R., J.U. Bowie & D. Eisenberg. 1992. Assessment of protein models with three-dimensional profiles. Nature 356: 83-85. http://dx.doi.org/10.1038/356083a0 [ Links ]
Messaoudi A., H. Belguith & J.B. Hamida. 2011. Three-Dimensional Structure of Arabidopsis thaliana Lipase Predicted by Homology Modeling Method. Evolutionary Bioinformatics 7: 99-105. http://dx.doi.org/10.4137/EBO.S7122 [ Links ]
Monsia T. & P. Monsia. 2013. A review on "microbial lipase-versatile tool for industrial applications". International Journal of Life Sciences Biotechnology and Pharma Research 2: 1-16. [ Links ]
Park I.H., S.H. Kim, Y.S. Lee, S.C. Lee, Y. Zhou, C.M. Kim, S.C. Ahn & Y.L. Choi. 2009. Gene Cloning, Purification, and Characterization of a Cold-Adapted Lipase Produced by Acinetobacter baumannii BD5. Journal of Microbiology and Biotechnology 19(2): 128–135. http://dx.doi.org/10.4014/jmb.0802.130 [ Links ]
Petersen T.N., S. Brunak, G. Heijne & H. Nielsen. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature Methods 8: 785-786. http://dx.doi.org/10.1038/nmeth.1701 [ Links ]
Ritchie D.W., D. Kozakov & S. Vajda. 2008. Accelerating and Focusing Protein-Protein Docking Correlations Using MultiDimensional Rotational FFT Generating Functions. Bioinformatics 24(17): 1865-1873. http://dx.doi.org/10.1093/bioinformatics/btn334 [ Links ]
Rost B. 1999. Twilight zone of protein sequence alignments. Protein Engineering 12: 85-94. https://doi.org/10.1093/protein/12.2.85 [ Links ]
Ruiz C., A. Blanco, F.I. Pastor & P. Diaz. 2002. Analysis of Bacillus megaterium lipolytic system and cloning of LipA, a novel subfamily I.4 bacterial lipase. FEMS Microbiology Letters 217(2): 263-267. http://dx.doi.org/10.1111/j.1574-6968.2002.tb11485.x [ Links ]
Saeed H.M., T.I. Zaghloul, A.I. Khalil & M.T. Abdelbaeth. 2006. Molecular cloning and expression in Escherichia coli of Pseudomonas aeruginosa lipase gene. Biotechnology 5(1): 62-68. http://dx.doi.org/10.3923/biotech.2006.62.68 [ Links ]
Sangeetha R., I. Arulpandi & A. Geetha. 2014. Molecular characterization of a proteolysis-resistant lipase from Bacillus pumilus SG2. Brazilian Journal of Microbiology 45(2): 389-393. http://dx.doi.org/10.1590/S1517-83822014000200004 [ Links ]
Schiraldi C., M. Giuliano & M. De Rosa. 2002. Perspectives on biotechnological applications of archaea. Archaea 1(2): 75-86. http://dx.doi.org/10.1155/2002/436561 [ Links ]
Sullivan E.R., J.G. Leahy & R.R. Colwell. 1999. Cloning and sequence analysis of the lipase and lipase chaperone-encoding genes from Acinetobacter calcoaceticus RAG-1, and redefinition of a Proteobacterial lipase family and an analogous lipase chaperone family. Gene 230(2): 277-285. https://doi.org/10.1016/S0378-1119(99)00026-8 [ Links ]
Tan Y. & K.J. Miller. 1992. Cloning, expression, and nucleotide sequence of a lipase gene from Pseudomonas fluorescens B52. Applied and Environmental Microbiology 58(4): 1402-1407. [ Links ]
Thompson J.D., T.J. Gibson, F. Plewniak, F. Jeanmougin & D.G. Higgins. 1997. The CLUSTALX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research 25(24): 4876-4882. http://dx.doi.org/10.1093/nar/25.24.4876 [ Links ]
Untergasser A., I. Cutcutache , T. Koressaar, J. Ye, B.C. Faircloth & M. Remm, S.G. Rozen. 2012. Primer3 new capabilities and interfaces. Nucleic Acids Research 40(15): e115. http://dx.doi.org/10.1093/nar/gks596 [ Links ]
Vivas-Reyes R., M. Ahumedo & J. Cabezas. 2010. Modelamiento por homología de la estructura tridimensional de la fosfolipasa A2 citosólica pancreática dependiente de calcio presente en Rattus norvegicus. Revista colombiana de química 39(2): 181-197. http://dx.doi.org/10.15446/rev.colomb.quim [ Links ]
Wang C., R. Guo, H. Yu & Y. Jia. 2008. Cloning and sequence analysis of a novel cold-adapted Lipase Gene from strain lip 35 (Pseudomonas sp.). Agricultural Sciences in China 7: 12161221. http://dx.doi.org/10.1016/S1671-2927(08)60167-4 [ Links ]
Wohlfarth S., C. Hoesche, C. Strunk & U.K. Winkler. 1992. Molecular genetics of the extracellular lipase of Pseudomonas aeruginosa PAO1. Journal of General Microbiology 138(7): 13251335. http://dx.doi.org/10.1099/00221287-138-7-1325 [ Links ]
Yang A.S. & B. Honig. 2000. An integrated approach to the analysis and modeling of protein sequences and structures. III. A comparative study of sequence conservation in protein structural families using multiple structural alignments. Journal of Molecular Biology 301: 665-711. http://dx.doi.org/10.1006/jmbi.2000.3975 [ Links ]
* Autor para correspondencia
Email hosep Avila: shonatan@hotmail.com
Email Amparo Iris Zavaleta: azavaletap@unmsm.edu.pe
Email Mercedes Palomino: mepa_8815@hotmail.com
Email Christian Solis Calero: c180276@dac.unicamp.br
Presentado: 27/11/2017
Aceptado: 12/09/2018
Publicado online: 25/09/2018

