











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La COVID-19 es una enfermedad causada por el virus SARS-CoV-2, un nuevo betacoronavirus que representa una grave amenaza para la salud a nivel mundial(1). Los primeros casos de COVID-19 se reportaron a finales de diciembre de 2019 en Wuhan-China, y tres meses después ya se había propagado por todo el mundo(1); por esta razón, la Organización Mundial de la Salud (OMS) la declaró pandemia el 11 de marzo del presente año1. Desde su aparición hasta el momento, las cifras de contagiados a nivel mundial superan los 36754395 casos confirmados y 1064838 fallecidos (10/10/2020)(2). De estas cifras, el Perú registra más de 843355 casos confirmados y 33158 fallecidos a la fecha (10/10/2020)3, siendo uno de los países con mayor número de muertes en Sudamérica y el mundo.
El 24 de enero del 2020, Zhu N. et al4publicaron la secuencia genética del SARS- CoV-2, mediante ensayo con PCR de transcripción inversa en tiempo real (RT-PCR), de pacientes con neumonía atípica de la ciudad de Wuhan. A raíz de este descubrimiento se determinó que el SARS CoV-2 presenta una región 5’ no traducible (UTR), un complejo replicasa (ORF1ab), genes S, E, M, N, una región 3’-UTR y otros marcos abiertos de lectura (ORFs) que difieren del resto de betacoronavirus4,5.
A pesar de ser un nuevo virus, el estudio del genoma y las proteínas de los coronavirus con los que comparte un gran porcentaje de identidad se ha desarrollado desde hace varios años atrás, principalmente investigaciones realizadas en SARS-CoV y MERS-CoV, lo que ha permitido sentar la base para el estudio de las proteínas del SARS-CoV-26,7.
Debido a la importancia de ciertas proteínas en procesos de replicación, transcripción y ensamblaje del SARS-CoV-2 se han realizado múltiples estudios con fármacos para su inhibición, como tratamiento del COVID-198; dentro de los cuales se encuentran la hidroxicloroquina, azitromicina y remdesivir que se han estudiado por mucho tiempo sin ningún resultado favorable8,9. Sin embargo, diversos investigadores han enfocado sus esfuerzos en experimentar con otras moléculas mediante estudios in silico, al ser más rápidos y baratos, para su posterior estudio in vivo o in vitro.
El presente trabajo busca profundizar desde el aspecto molecular la patogénesis del COVID-19, mediante la descripción del rol de las proteínas no estructurales (Nsps) y estructurales del SARS-CoV-24en la infección de sus células diana. Asimismo, dar a conocer posibles tratamientos farmacológicos de estudios in silico que tienen como objetivo la inhibición de proteínas esenciales en el desarrollo del ciclo viral y podrían servir de base para posteriores estudios tanto in vitro como in vivo.
El procedimiento para la búsqueda de información se inició buscando todas las proteínas no estructurales (Nsps) y las proteínas estructurales del SARS-CoV-2 en MeSH. Una vez identificadas se hizo la búsqueda de cada una de ellas hasta el 6 de setiembre en las bases de datos PubMed, Scielo, Google Scholar y LinkSpringer, empleando como criterios publicación en revistas indexadas, que respondan a los objetivos y que tengan información relevante. Se encontraron 100 artículos y se seleccionaron 68, la mayoría de PubMed; solo 4 tuvieron la excepción de ser preprint por la relevancia de su información.
Para la elaboración de lasFigura 2yFigura 3, primero se obtuvo los códigos de identificación de cada proteína del Protein Data Bank(10)(PDB), luego se visualizó su secuencia aminoacídica con el programa Jalview(11)y con su extensión Quimera12se visualizó la estructura 3D de cada proteína. Se utilizó el programa CellPAINT13para dibujar el virus, la célula y las proteínas estructurales del SARS-CoV-2 publicadas en laFigura 4conforme lo señalan los autores(13). Asimismo, se utilizó el programa BioRender(14)empleando sus figuras prediseñadas de la célula y organelas, añadiendo la estructura del virus y sus proteínas para esquematizar el proceso de patogénesis del SARS-CoV-2 de laFigura 5 14.
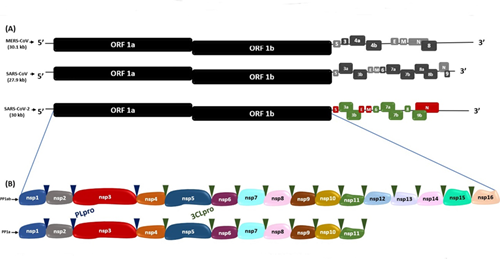
Figura 1. Secuencias genómicas de SARS-CoV-2, MERS-CoV y SARS-CoV. (A) Comparación de secuencia genómica de SARS-CoV-2 con MERS-CoV y SARS-CoV. (B) Poliproteína 1ab (PP1ab) y poliproteína 1a (PP1a) codificadas por ORF 1a y ORF 1b, con sus respectivas proteínas no estructurales (Nsps). Los triángulos azul y verde indican los sitios de escisión de la proteasa PLpro y 3CLpro, respectivamente.
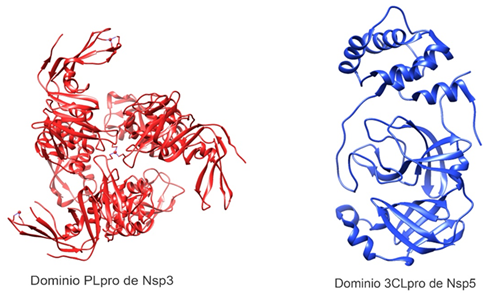
Figura 2. Estructura tridimensional de proteasas del SARS-CoV-2. Dominio PLpro (papain-like protease) de Nsp3 (PDB: 6W9C) y Nsp5 (PDB: 6Y2E) con su dominio 3CLpro (3-chymotrypsin-like protease). Elaborado con UCSF Chimera--a visualization system for exploratory research and analysis. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004 Oct;25(13):1605-12.
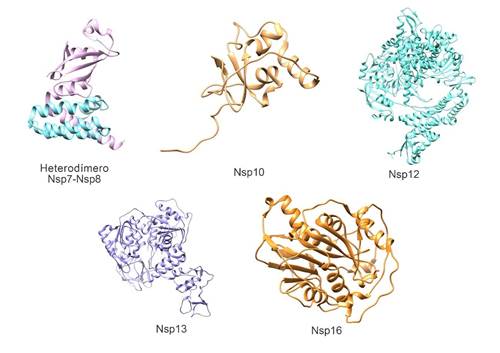
Figura 3. Estructura tridimensional de proteínas del complejo replicasa-transcriptasa del SARS-CoV-2. Heterodímero Nsp7-Nsp8 (Nsp7 de color turquesa, Nsp8 de color magenta) (PDB: 7JLT), Nsp10 (PDB: 6W75), Nsp12 (PDB: 7BW4), Nsp13 (PDB: 6ZSL) y Nsp16 (PDB: 6W75). Elaborado con UCSF Chimera--a visualization system for exploratory research and analysis. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J Comput Chem. 2004 Oct;25(13):1605-12.
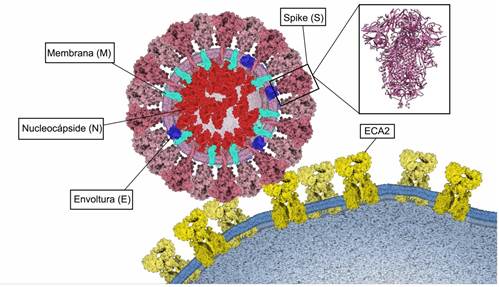
Figura 4. Unión del SARS-CoV-2 a ECA2. Proteínas estructurales Spike (S) de color rosado, Membrana (M) de color turquesa, Envoltura (E) de color azul y Nucleocápside (N) de color rojo. Estructura tridimensional de la proteína Spike (PDB: 6VXX) y su unión a ECA2 (color amarillo) en la membrana de la célula huésped. Elaborado con Gardner A, Autin L, Barbaro BA, Olson AJ, Goodsell DS. CellPAINT: Interactive Illustration of Dynamic Mesoscale Cellular Environments. IEEE Comput Graph Appl. 2018;38(6):51-66.
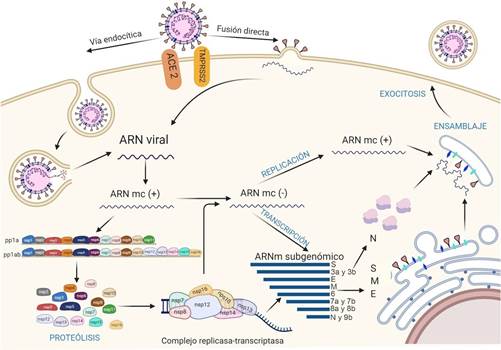
Figura 5. Mecanismo de interacción y patogénesis del SARS-CoV-2. Entrada del SARS-CoV-2 a través de 2 vías: endocitosis (a la izquierda) y fusión directa (a la derecha). El ARN genómico ingresa a la célula y el ARN monocatenario en sentido positivo (ARN mc +) se traduce en las poliproteínas pp1a y pp1ab a partir de las regiones ORF 1a y ORF 1ab. Posteriormente, un proceso de autoclivaje por 3CLpro y Mpro, dará lugar a las 16 proteínas no estructurales (Nsps), que formarán el complejo replicasa transcriptasa (RTC), que producirá ARN monocatenario de polaridad negativa a partir de la cadena positiva; que se asociará con la proteína de nucleocápside. Por otra parte, el complejo RTC, sintetizará ARN subgenómico (ARN sg) que codificarán las proteínas S, M y E, ensambladas en retículo endoplasmático antes de ser transportadas al compartimiento RE-Golgi, donde se asociará con el nuevo ARN genómico y la proteína N. Finalmente se exportará en forma de vesículas para la posterior liberación del nuevo virus. Elaborado con https://biorender.com/
ORGANIZACIÓN GENÓMICA DEL SARS-CoV-2
El SARS-CoV-2 pertenece al género de los betacoronavirus y comparte una identidad de secuencia genómica del 79,6% con SARS-CoV y del 50% con el MERS-CoV(5). Tiene un ARN genómico monocatenario de sentido positivo (ssARN) con una longitud de aproximadamente 30 Kb6; además, presenta una cola poliadenilada (poli-A) en el extremo 3’ y un capuchón metilado en el extremo 5’, teniendo una similitud estructuralmente al ARN mensajero (ARNm) de células eucariotas(15).
Este ARN consta de 15 marcos abiertos de lectura (ORFs) que son secuencias de ARN comprendida entre un codón de inicio de la traducción y un codón de terminación(16). En SARS-CoV, MERS-CoV y SARS-CoV-216, los dos tercios próximos al extremo 5´- terminal de su genoma, se encuentran los ORF1a y ORF1b, que codifican las poliproteínas 1a (PP1a) y 1ab (PP1ab), respectivamente.El clivaje de estas poliproteínas origina a las proteínas no estructurales (Nsp1-16), que conforman el complejo viral replicasa-transcriptasa16. Por otro lado, en el tercio próximo al extremo 3’, se encuentran los ORFs S, E, M y N, que codifican a las proteínas spike (S), envoltura (E), nucleocápside (N) y membrana (M), respectivamente, y los ORFs 3a, 3b, 6, 7a, 7b, 8, 9a, 9b y 10 que codifican a las proteínas accesorias16,17(Figura 1).
PROTEÍNAS DEL SARS-CoV-2
El conocimiento de las proteínas y sus funciones en el SARS-CoV-2 se ha obtenido principalmente de diversas investigaciones sobre el SARS-CoV y otros betacoronavirus relacionados, como el MERS-CoV, pero en menor proporción, debido a las similitudes del genoma del SARS-CoV-2 con el de este grupo de virus5,18. En laTabla 1se muestran los porcentajes de identidad de las proteínas del SARS-CoV-2 con sus homólogas del SARS-CoV6,16,19-24.
Tabla 1. Porcentaje de identidad entre proteínas de SARS-CoV-2 y SARS-CoV.
| Proteína | % Identidad con SARS-CoV |
|---|---|
| Nsp1 | 86% |
| Nsp2 | <70% |
| Nsp3 | 76% |
| Nsp4 | 80% |
| Nsp5 | 97% |
| Nsp6 | 88% |
| Nsp7 | 67% |
| Nsp8 | 85% |
| Nsp9 | 97% |
| Nsp10 | 99% |
| Nsp12 | 97% |
| Nsp13 | 100% |
| Nsp14 | 95% |
| Nsp15 | 88% |
| Nsp16 | 94% |
| Spike | 75% |
| Envoltura | 89% |
| Membrana | 90.50% |
| Nucleocápside | 90.52% |
El SARS-CoV-2 cuenta con 16 proteínas no estructurales, cuatro proteínas estructurales y ocho proteínas accesorias16, las cuales se describirán a continuación.
Proteínas no estructurales
Las 16 proteínas no estructurales provienen del clivaje proteolítico de las poliproteínas PP1a y PP1ab expresada por ORF1a y ORF1ab16, respectivamente. Estas cumplen un rol fundamental en la replicación del virus dentro de las células huésped y algunas de ellas son el objetivo de diversos fármacos en desarrollo7,25.
Nsp1
En su estructura posee 6 láminas beta que conforman un barril con una hélice alfa cubriendo una parte terminal del barril y otra a lo largo de este26.
Esta proteína interactúa con la subunidad ribosomal 40S de la célula huésped por medio de los residuos de Lys164 e His165, a través de esta subunidad conseguirá el acceso al ARNm del huésped27. El Nsp1 no permite la unión de la subunidad ribosomal 40S con la subunidad 60S, inhibiendo así la traducción del ARNm27. Después, recluta a una endonucleasa que induce a un clivaje endonucleótico en el extremo 5’-UTR del ARNm; esto conlleva a una degradación por la exoribonucleasa Xrn1 del extremo 5’ truncado27.
Nsp2
Presenta una estructura estabilizada por Gln321, debido a la longitud de su cadena lateral, polaridad y potencial para formar enlaces de hidrógeno28. Esta se une a las proteínas PHB 1 y PHB2 (prohibitin1 y 2) de la célula huésped, que participan en el progreso del ciclo celular, migración celular, diferenciación celular, apoptosis y biogénesis mitocondrial28. Sin embargo, el mecanismo de unión de Nsp2 a las proteínas PHB1 y PHB2 aún no está estudiado18.
Nsp3
Esta es la proteína más grande del SARS-CoV-2 y a partir del extremo N-terminal, presenta de forma secuencial un dominio similar a la ubiquitina que se une al ARN monocatenario, a un módulo de unión a ribosa ADP, a un dominio de unión de poli (A) trenzado, a una proteasa viral de tipo papaína, a un dominio de unión a ácido nucleico y a un receptor C acoplado a proteína G29.
Esta proteína junto con Nsp4 y Nsp6, regula el sitio de replicación reclutando la proteína replicasa a la membrana del huésped28. Los dominios transmembrana de múltiples capas de la proteína Nsp3 sirven como un andamio para el ensamblaje del complejo replicasa-transcriptasa asociado a la membrana30.
El dominio proteasa similar a la papaína (PLpro) del Nsp3 (Figura 2) es responsable de la liberación de Nsp1, Nsp2 y Nsp3 de la región N-terminal de las poliproteínas 1a y 1ab29. Además, este dominio se puede unir a las proteínas RIG-I, NEMO, TRAF6 para que no activen los factores de transcripción IRF3 y NFKB, quienes coordinan la expresión de los interferones tipo I.Debido a ello, se bloquea la producción de citocinas importantes involucradas en la activación de la respuesta inmune innata del huésped contra la infección viral29,31.
Nsp4
Esta proteína transmembrana presenta múltiples sustituciones cerca de su región N-terminal y tiene un C-terminal bastante conservado; estas regiones son citosólicas18. Además, posee un dominio muy conservado similar a las defensinas humanas involucradas en la inmunidad innata que está constituido por residuos de aminoácidos que se extienden desde las posiciones 217 a 23732.
La coexpresión de Nsp4 con el tercio C-terminal de Nsp3 se produce en las posiciones 112-164 en su bucle luminal, permitiendo así la redistribución del retículo endoplasmático a la región perinuclear para la inducción de la formación de vesículas de doble membrana, pero cuando se expresa individualmente la Nsp4 se localiza en el retículo endoplasmático33. Además , al expresarse junto con la Nsp6 permiten una replicación óptima dentro de las células huésped. Por otro lado, se ha observado que en los residuos de aminoácidos His120 y Phe121 en SARS-CoV, Nsp4 juega papeles cruciales en la remodelación de la membrana a través de su interacción con Nsp333.
Nsp5
La proteasa principal de SARS-CoV-2 (Mpro, NSP5, 3CLpro ) es una cisteína proteasa homodimérica de 67,6 kDa altamente conservada, que difiere solo en 12 aminoácidos con la correspondiente proteasa M pro del SARS-CoV20.
Está conformada por los dominios I (residuos 8-101), II (residuos 102-184) y III (residuos 201-303), con un bucle largo (residuos 185-200) que conecta los dominios II y III20(Figura 2). El residuo Glu166 es un aminoácido clave implicado en la dimerización de Mpro y en la creación de bolsillo de unión al sustrato34. Además, los residuos Cys141 e His41 forman una díada catalítica en el sitio activo de la proteína, esencial para su función20,34,35.
Esta proteasa se escinde de las poliproteínas para producir enzimas maduras, y luego escinde más proteínas no estructurales downstream en 11 sitios para liberar Nsp4 - Nsp16. Además, actúa como mediador en la maduración de Nsps, que es esencial en el ciclo de vida del virus34,36.
Nsp6
Proteína transmembrana que consta de 290 aminoácidos y se localiza en el retículo endoplasmático37. La parte de su estructura que se encuentra en la región de la membrana externa posee múltiples residuos de fenilalanina, lo cual favorece la afinidad de esta proteína con la membrana del retículo y haría su unión más estable38.
El Nsp6 forma complejos con Nsp3 y Nsp4; además, está implicada en la formación de vesículas de doble membrana derivadas de retículo endoplásmico durante la replicación del coronavirus21,37. Se hipotetiza que la formación de estas vesículas de doble membrana estan inducidas como omegasomas por la inhibición de la diana de rapamicina en células de mamífero (mTOR) por Nsp6 o bien por la activación de la autofagia a través de inducción de vías alternas, las cuales continúan hasta la formación del autofagosoma donde, en condiciones normales, pasaría a formar un autolisosoma para degradar su contenido, en este caso viral, por la infección de SARS-CoV-221,37. Sin embargo, este proceso no se daría gracias a la Nsp6, ya que formaría autofagosomas más pequeños (menos de 0.5um) de lo normal (aprox. 1um). Se sugiere que el tamaño reducido de estos autofagosomas limita su capacidad de fusionarse con los lisosomas, esto beneficiaría la replicación viral al prevenir la maduración de las vesículas endosómicas y autofágicas y, por consecuencia, su capacidad de degradar los elementos virales, proporcionándoles una nueva maquinaria para su replicación en condiciones seguras21,37.
Nsp7 y Nsp8
Tanto la estructura de Nsp7 como de Nsp8 es predominantemente alfa helicoidal38,39(Figura 3). Ambas forman un super complejo hexadecamérico que adopta una estructura cilíndrica hueca y puede participar en la replicación viral actuando como primasa, cuyas propiedades electrostáticas positivas implican que confiere procesividad a la ARN polimerasa dependiente de ARN40.
Nsp9
Presenta un núcleo conformado por un pequeño barril β cerrado de siete hebras, desde el cual se proyecta una serie de bucles extendidos hacia afuera22. Los bucles alargados unen las cadenas β individuales del barril, junto con una cadena β N-terminal en proyección y la hélice α1 C-terminal; estos dos últimos elementos constituyen los componentes principales en la disposición dimérica de la proteína22,41. Existen dos formas para la dimerización de la Nsp9, una es la interacción que se da entre las hélices α paralelas de cada monómero que contienen el motivo de interacción proteína-proteína Gli-X-X-X-Gli; y la otra forma es mediante una interfaz de lámina beta estabilizada por interacciones de átomos de la cadena principal dentro de las regiones de lámina de cada monómero41.
El Nsp9 tiene capacidad de unión a ARN y ADN, por lo que media la replicación viral, la virulencia general y la reproducción viral del ARN genómico; siendo, probablemente, un miembro del complejo de replicación22.
Nsp10
La estructura de esta pequeña proteína de 139 aminoácidos (Figura 3) está conformada por un par de láminas beta antiparalelas en el centro, rodeadas en un lado por un bucle grande que las cruza y por 5 hélices alfa cuyos bucles forman dos dominios de dedos de zinc, el primer sitio de unión a zinc está coordinado por los residuos Cys74, Cys77, His83 y Cys90 y el segundo sitio de enlace a zinc está coordinado por Cys117, Cys120, Cys128 y Cys130(16, 42). La función de estos sitios en otros coronavirus está involucrada con la unión no específica al ARN42.
El dominio C-terminal de Nsp10 forma múltiples interacciones con el dominio ExoN de Nsp14, lo que afecta fuertemente su actividad nucleolítica, la cual mejora hasta 35 veces43. Por otra parte, esta proteína también actúa como cofactor de Nsp16, aumentando la actividad de su dominio 2’-O-MTasa43.
Nsp12
En su estructura encontramos un dominio β hairpin conformado por una horquilla β N-terminal en los residuos 31-50, un dominio NiRAN (nidovirus RdRp-associated nucleotidyl-transferase) en los residuos 115-250 con siete hélices y 3 láminas β, un dominio interface en los residuos 251-365 compuesto por tres hélices y cinco láminas β que conecta el dominioNiRANy el dominio RdRp, el cual tiene una configuración ahuecada, con subdominios fingers en los residuos 397-581 y 621-679, un subdominiothumben los residuos 819-920, y un subdominiopalm, los cuales forman un círculo cerrado44(Figura 3).
El dominio RdRp tiene actividad ARN polimerasa dependiente de ARN, pero por sí solo presenta baja actividad; por ello requiere factores accesorios, que son las proteínas Nsp7 y Nsp8 con los que conforma un complejo, el cual presenta motivos con residuos conservados de unión a Zn2+ en Cys487 His642, Cys645, Cys646 y en His295, Cys301, Cys306, Cys31044. Esto incrementa la unión de RdRp al molde-primer de ARN40,44.
Nsp13
En su estructura presenta cinco dominios: dominio en cintas - ZBD, dominio tallo, dominio 1b, dominio 1a, dominio 2a y un sitio activo NTPasa que está compuesto por Lys288, Ser289, Asp374, Glu375, Gln404 y Arg567; estos son vitales para el desenrollado de una molécula de ARN dúplex45(Figura 3).
A concentraciones elevadas de ATP, la actividad helicasa de la proteína presenta una mayor afinidad por el ARN dúplex, que se desarrolla en tres pasos: Primero, el Nsp13 se une a la cola 5′-ss en presencia de ATP sin hidrólisis de ATP; luego, al agregar iones de magnesio desencadena la hidrólisis de ATP y finalmente, el Nsp13 permite separar el ARN dúplex y se transloca a lo largo del ARN desenrollado en dirección 5´a 3´46.
Nsp14
Esta proteína posee un dominio N-terminal (ExoN) que incluye tres motivos (I(DE), II(E), III(D)) y un dominio carboxiterminal que contiene (N7 guanina)-metil transferasa (N7-MTasa)47. El dominio ExoN presenta un pliegue alfa/beta, compuesta por una lámina beta central formada por cinco filamentos beta flanqueados por hélices alfa, a excepción de la cadena beta 3, y sus residuos catalíticos incluyen Asp90, Glu92 Glu191, Asp27247. Por otra parte, el dominio N7-MTasa está formado por una lámina beta constituido por cinco cadenas beta y un motivo canónico de unión a S-adenosilmetionina (SAM)47. Una región de bisagra separa el dominio ExoN del dominio N7-MTasa, esta es flexible y consta de un bucle y tres hebras, lo que permite movimientos laterales y rotacionales de los dos dominios para coordinar las actividades enzimáticas43,47.
El Nsp14 funciona como una metiltransferasa dependiente de S-adenosil metionina (SAM) (guanina-N7) (N7-MTasa)47. Después de la hidrólisis del ARN naciente por el Nsp13, se forma ARN-pp. Una guanililtransferasa (GTasa) desconocida hidroliza GTP, transfiere el producto GMP a ARN-pp y crea ARN-Gppp. Luego, SAM metila la guanina 5' del ARN-Gppp en la posición N7, formando la tapa 5’(m7GppN-ARN)43,47.
Nsp15
Esta proteína es una endoribonucleasa específica de uridilato de ARN nidovírico (NendoU) con unidades monoméricas compuestas por 345 aminoácidos que se pliegan en tres dominios48. El dominio N-terminal se compone de una hoja-β antiparalela envuelta alrededor de dos α -hélices (α 1 y α 2) y el dominio medio subsiguiente está formado por diez filamentos β organizados en tres pines, una hoja-βmixta, y tres hélices cortas(48). El dominio catalítico NendoU del terminal C contiene dos láminas β antiparalelas con bordes que alojan un sitio catalítico48.
El sitio activo está ubicado en un surco poco profundo entre las dos hojasβ, lleva seis residuos clave: His235, His250, Lys290, Thr341, Tyr343 y Ser294, de los cuales, los residuos His235, His250 y Lys290 constituyen la tríada catalítica, mientras que Ser294 y Tyr343 se encargan de la especificidad de NendoU48,49
Nsp16
En su estructura contiene una tétrada catalítica altamente conservada (Lys-Asp-Lys-Glu), distintiva de las ARN 2′-O-MTasas, dentro de un núcleo compuesto por un pliegue de lámina β tipo Rossmann decorado por once hélices α, siete cadenas β y bucles50. Además, forma el pliegue del SARS-CoV-2, conformado por una lámina β la cual está encajonada por bucles y hélices α(50)(Figura 3).
Proteínas estructurales
Se han identificado cuatro proteínas estructurales en el SARS-CoV-2, las cuales también están presentes en otros coronavirus. Estas son las proteínas Spike(S), Envoltura(E), Membrana(M) y Nucleocápside(N) (Figura 4).
Proteína Spike(S)
Esta proteína tiene un peso molecular de 180 kDa16. Su estructura contiene las subunidades funcionales S1 y S2, ubicadas en su ectodominio51.
La subunidad S1 presenta un dominio N-terminal, un dominio C-terminal51y un dominio de unión al receptor conservado (RBD) que contiene un núcleo y un motivo de unión al receptor (RBM)51. Esta subunidad media la unión al receptor ECA 2, donde los residuos de aminoácidos como Lys317 y Phe486 del dominio RBD podrían ser claves para esta interacción51,52.
Por otro lado, la subunidad S2 posee en su estructura un dominio péptido de fusión (FP), dominios de repetición heptad-1 y -2 (HR1, HR2) y un dominio transmembrana (TM), que le permiten la fusión de las membranas viral y celular51,52.
La proteína S requiere un clivaje por proteasa para la activación de su potencial de fusión. Se han propuesto dos pasos secuenciales para el modelo de clivaje; un clivaje inicial entre S1 y S2, y la posterior activación de clivaje en el sitio S2’53.
Además, presenta una gran superficie mutada, con cuatro insertos nuevos en la proteína; de los cuales, tres se ubican en el primer dominio NTD, mientras que el cuarto se ubica inmediatamente antes del sitio de escisión S2 y dentro de la interfaz de interacción de homo-trimerización16. Asimismo, el dominio RBD no está afectado por estos insertos, pero es la región más mutada con potenciales alteraciones en su función de unión a ECA216.
Proteína de Envoltura(E)
Presenta gran identidad con las secuencias de otros coronavirus; sin embargo, existen características distintivas como la sustitución de residuos de glutamato, glutamina o aspartato por arginina en la posición y el reemplazo de la diada Ser-Phe por Thr-Val en las posiciones 55-5654.
Esta proteína es la más pequeña de las cuatro proteínas estructurales, con 76 aminoácidos de longitud55,56. Su estructura posee un extremo amino hidrófilo corto cargado negativamente que consta de 7 a 12 aminoácidos, seguido de un dominio transmembrana hidrófobo (TMD) grande de 25 aminoácidos, y termina con un extremo carboxilo hidrófilo largo de carga variable55,56. La región hidrófoba de la TMD contiene una hélice α anfipática que se oligomeriza para formar un poro conductor de iones en las membranas;una parte del TMD consta de dos aminoácidos neutros no polares, Val y Leu, que confieren una fuerte hidrofobicidad a la proteína(56). El extremo C-terminal también exhibe cierta hidrofobicidad, pero menos que el TMD debido a la presencia de un grupo de aminoácidos básicos cargados positivamente, además contiene un residuo de prolina conservado centrado en un motivo β-coil-β, que probablemente funciona como una señal de dirección hacia el complejo de Golgi56.
Proteína de Membrana(M)
Esta glicoproteína integral de membrana es la mayoritaria y proporciona la morfología al virión57,58. Posee una longitud de aproximadamente 220-260 aminoácidos con un dominio N-terminal de longitud corta, integrada en la membrana del virus por medio de tres dominios transmembrana etiquetados como tm1, tm2 y tm3(57-59). Su extremo terminal amino corto glicosilado constituye un ectodominio fuera de la membrana, mientras su endodominio C-terminal se sitúa en el lado citoplasmático de la membrana del virión57,58. El ectodominio puede ser glicosilado, afectando el tropismo de los órganos a infectar y la capacidad inductora de interferón (IFN) de algunos coronavirus58,59. Además, presenta la inserción de un residuo de serina en la posición 4 como característica única en el SARS-CoV-223.
Durante el ensamblaje, proporciona un andamio para las partículas virales, estabiliza a la proteína N (complejo proteína N - ARN) y al núcleo interno de los viriones; además es necesaria para la retención de la proteína S en el compartimento intermedio ER-Golgi (ERGIC) y su incorporación a nuevos viriones57. La coexpresión de M y E forman la envoltura viral, su interacción es suficiente para la producción y liberación de partículas similares a virus (VLP)58,59.
Proteína Nucleocápside(N)
Su estructura está conformada por dos dominios bien plegados, conocidos como dominio N-terminal (NTD) y dominio C-terminal (CTD)24, ambos dominios son ricos en cadenas β, pero CTD tiene además algunas hélices cortas24.
Se une directamente al ARN viral y le provee estabilidad60. Además, se ha encontrado que antagoniza al ARNi antiviral e inhibe la actividad del complejo ciclina-CDK (cyclin-cyclin-dependent kinase); esta inactivación resulta en la hidrofosforilación de la proteína retinoblastoma y a su vez inhibe la progresión de la fase S en el ciclo celular18.
Proteínas accesorias
Las proteínas accesorias del SARS-CoV-2 son expresadas por los genes ORF3a, ORF3b, ORF6, ORF7a, ORF7b, ORF8, ORF9a, ORF9b y ORF10. Varias de estas proteínas tienen funciones aún desconocidas, se sospecha que no intervienen en la replicación viral pero pueden tener roles importantes en la patogénesis viral18.
PATOGÉNESIS DEL COVID-19
Entrada del virus y unión con receptor ECA2
El SARS-CoV-2 puede ingresar a la célula huésped a través de dos rutas: endocitosis o fusión directa con la superficie celular15,51. Para la ruta de endocitosis, el virus es encapsulado por el endosoma después de la unión a su receptor ECA215. Luego, el ambiente de pH bajo promueve la escisión de la proteína S con la catepsina L (CPL) de cisteína proteasa dependiente del pH50. Por otra parte, en la ruta de fusión directa con la superficie celular, después de la unión del dominio RBD de la subunidad S1 al receptor ECA2, la proteasa transmembrana serina 2 (TMPRSS2) escinde y activa a la proteína S, en el ectodominio conformado por S1 y S2, que permite la fusión de la membrana viral con la membrana de la célula huésped15,51,61.
La activación de la proteína S se realiza en el sitio de escisión S1/S2 también llamado "sitio polibásico" o "sitio multibásico", el cual contiene varios residuos de arginina (Ser-Pro-Arg-Arg-Ala-Arg ↓ Ser)15,61. También se necesita un segundo corte en S2′ en Lys-Arg ↓ Ser-Phe53, el cual no difiere de otros virus similares al SARS-CoV-2, lo que activa el péptido de fusión (FP) que une las membranas del huésped y el virus, constituyendo la etapa intermedia de fusión53,62,63. Por último, el segmento entre HR1 y HR2 cambia en su conformación, se doblan y forman un heptámero (6-HB) que une ambas membranas facilitando el ingreso del virus61.
También se tiene conocimiento que otras enzimas pro proteína convertasas, como la furina o tripsina de la célula huésped, reconocen el sitio de escisión S1/S261.
Mecanismo de replicación, transcripción y traducción del SARS-CoV-2
Después del ingreso del virus a la célula huésped, por la vía endocítica, se requiere acidificación endosómica, es decir una disminución de pH mediada por lisosomas(64), permitiendo la unión de la membrana del endosoma con la envoltura viral, liberando su nucleocápside al citoplasma64,65.
El ARN del virus en las regiones ORF1a y ORF1b actúa como un ARNm donde se transcribe directamente el gen de la replicasa viral con la maquinaria de la célula huésped, traduciéndose en las poliproteínas PP1a y PP1ab15. Estas se escinden por las proteasas tipo papaína (PLpro; correspondiente a Nsp3) y tipo 3C (3CLpro o Mpro; correspondiente a Nsp5) para formar las 16 proteínas no estructurales (Nsps)15,64.
A continuación, las proteínas no estructurales se reorganizan en vesículas de doble membrana a partir del retículo endoplasmático (RE)64, y se ensamblan en la región perinuclear, en el complejo replicasa-transcriptasa (RTC), creando un entorno adecuado para la síntesis de ARN de sentido negativo (-) a través de la replicación y la transcripción, de manera que replica y sintetiza un conjunto de ARNm subgenómicos (sgARN)64,65.
Los ARN subgenómicos se sintetizan combinando longitudes variables del extremo 3' del genoma con la secuencia líder 5' necesaria para la traducción. Estos ARN subgenómicos (-) se transcriben en ARNm subgenómicos (+), que codifican a las proteínas estructurales S, M, E, N y las accesorias (hacia el extremo 3’)64,65.
El ARN genómico viral recién sintetizado se une a la proteína N originando la nucleocápside64. Las proteínas S, M, E y las accesorias, expresadas a partir de los sgARN, son sintetizadas en las membranas del RE y luego transportadas al complejo de Golgi para ser ensambladas con la nucleocápside, produciendo nuevas partículas víricas; que por medio del sistema de transporte de vesículas, viajarán hasta la superficie, liberándolo por exocitosis(64,65)(Figura 5).
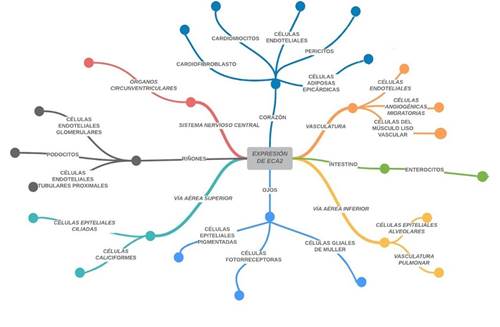
Como ya se mencionó anteriormente, el SARS-CoV-2 utiliza como receptor a la ECA2, la cual está expresada en diferentes células del sistema vascular, sistema nervioso central, ojos, vías aéreas superiores, corazón, pulmones e intestino; siendo más vulnerables los últimos tres66(Figura 6).
ESTUDIOS IN SILICO DE POSIBLES TRATAMIENTOS FARMACOLÓGICOS
La expresión in silico se utiliza para caracterizar aquellos estudios que son completamente ejecutados utilizando modelos de computadora. Significa que el entorno y el comportamiento del objeto de estudio bajo este medio están representados por modelos que simulan sus características relevantes a través de ingeniería de software. La ventaja de estos estudios es la rapidez de ejecución, el bajo costo y la capacidad de reducir el uso de animales; por ello, su uso ha sido una estrategia para acelerar el descubrimiento de nuevos fármacos potenciales como los que se describen en esta sección67. El diseño de prototipos de fármacos in silico, en los estudios citados en este artículo, abarca principalmente la relación estructura-actividad, en este caso entre las proteínas del SARS-CoV-2 y las moléculas de los fármacos67.
Las proteínas como unidades funcionales de la célula representan un sitio principal de acción farmacológica y xenobiótica(68). Por lo tanto, caracterizar las proteínas objetivo es fundamental para conocer el mecanismo de acción de los fármacos y los xenobióticos. El acoplamiento de sustratos en el sitio activo de una proteína de estructura conocida o modelada se puede utilizar para explicar las interacciones proteína-sustrato y posiblemente predecir las sustancias químicas que interactuarán con las proteínas69.
Comprender cómo los diferentes aminoácidos interactúan con los sustratos (ligando, proteína, fármaco, etc.) es crucial para explicar la conformación y la afinidad de las interacciones proteína-ligando. Esta técnica, también conocida como diseño de fármacos basado en la estructura, utiliza datos de estructura de proteínas 3D existentes para predecir las interacciones proteína-ligando utilizando softwares de acoplamiento (Docking) de ligando asistido por computadora69. El Docking usa una función de puntuación basada en la energía, las puntuaciones de energía más bajas representan mejores enlaces proteína-ligando en comparación con los valores de energía más altos70. La función de puntuación se define por la suma de la energía de interacción ligando-proteína la energía interna del ligando70.
Las moléculas que se han encontrado en los diversos estudios in silico realizados se han dividido en dos grupos: aquellas que son medicamentos aprobados para el uso humano que se intentan repropositar para tratar el SARS-CoV-2(71-73)(Tabla 1) y aquellas de origen vegetal(74-76)(Tabla 2).
Tabla 2. Medicamentos aprobados para el uso humano que se intentan repropositar para tratar el SARS-CoV-2.
| Autor, Año, Número de Referencia | Molécula | Efecto esperado en la estructura del SARS-CoV-2 | Resultado esperado | Uso común en medicina |
|---|---|---|---|---|
| Sarma, P; et al. 2020 (71) | 8- (2-hidroxietil) aminofilina | Unión al dominio N-terminal de la proteína N | Inhibición de la replicación | Broncodilatador |
| Sarma, P; et al. 2020 (71) | etil (4S) -4-metil-2-oxo-6 - ((1S) -1-feniletil) -3,4-dihidro-1H-pirimidina-5-carboxilato | Unión al dominio N-terminal de la proteína N | Inhibición de la replicación | Antiviral |
| Lobo-Galo, N; et al. 2020 (72) | Disulfiram | Inhibición de la proteasa viral | Inhibición de la replicación | Alcoholismo |
| Mahanta, S; et al. 2020 (73) | Viomycin | Atacar en el sitio activo de la Mpro | Inhibición de la replicación | Tuberculosis |
CONCLUSIONES
Las Nsps participan en el ciclo viral del virus y favorecen su infección; mientras que las proteínas estructurales median la unión y fusión del virus a la célula huésped, así como su ensamblaje; hasta el momento se desconoce las funciones de las proteínas accesorias. La activación de la proteína S por TMPRSS2, furina, tripsina o catepsinas permite el ingreso del SARS-CoV-2 a la célula huésped; en donde su ARN viral traduce las proteínas del complejo replicasa-transcriptasa, el cual inicia la síntesis de un molde para los ARN genómicos nuevos, quienes traducen las proteínas estructurales. Estudios in silico de muchas moléculas re propositadas y de origen vegetal han mostrado su acción inhibitoria en las proteínas Nsp3, Nsp5, N, M y E del SARS-CoV-2.
Tabla 3. Moléculas de origen natural aptas para el uso humano para tratamiento contra COVID-19.
| Autor, Año, Número de Referencia. | Molécula | Efecto esperado en la estructura del SARS-CoV-2 | Resultado | Fuente de obtención |
|---|---|---|---|---|
| Borkotoky, S; et al. 2020 (74) | Nimbolin A | Inhibición de los canales iónicos de la proteína E e interactúa principalmente con el dominio transmembrana de la proteína M | Previene la propagación del virus | Azadirachta indica (Neem) |
| Borkotoky, S; et al. 2020 (74) | Nimocin | Inhibición de los canales iónicos de la proteína E e interactúa principalmente con el dominio transmembrana de la proteína M | Pueden dificultar el ensamblaje del virus | Azadirachta indica (Neem) |
| Borkotoky, S; et al. 2020 (74) | 7-Deacetyl-7-Benzoylgedunin | Inhibición de los canales iónicos de la proteína E | Previene la propagación del virus | Azadirachta indica (Neem) |
| Borkotoky, S; et al. 2020 (74) | 24-Methylenecycloartanol | Inhibición de los canales iónicos de la proteína E | Previene la propagación del virus | Azadirachta indica (Neem) |
| Borkotoky, S; et al. 2020 (74) | Cycloeucalenone | Inhibición de los canales iónicos de la proteína E | Previene la propagación del virus | Azadirachta indica (Neem) |
| Borkotoky, S; et al. 2020 (74) | Phytosterol | Interactúan con la región C-terminal, principalmente con el dominio conservado CD de la proteína M | Pueden dificultar el ensamblaje del virus | Azadirachta indica (Neem) |
| Borkotoky, S; et al. 2020 (74) | Beta-Amyrin | Interactúan con la región C-terminal, principalmente con el dominio conservado CD de la proteínas M | Pueden dificultar el ensamblaje del virus | Azadirachta indica (Neem) |
| Borkotoky, S; et al. 2020 (74) | 24-Methylenecycloartan-3-one | Interactúan con la región C-terminal, principalmente con el dominio conservado CD de la proteína M | Pueden dificultar el ensamblaje del virus | Azadirachta indica (Neem) |
| Bhardwaj, VK; et al. 2020 (75) | Oolonghomobisflavan-A | Interactúan con el sitio activo de la Mpro uniéndose a residuos críticos. | Inhibición de la replicación | Planta de té |
| Bhardwaj, VK; et al. 2020 (75) | Theasinensin-D | Interactúan con el sitio activo de la Mpro uniéndose a residuos críticos. | Inhibición de la replicación | Planta de té |
| Bhardwaj, VK; et al. 2020 (75) | Theaflavin-3-O-gallate | Interactúan con el sitio activo de la Mpro uniéndose a residuos críticos | Inhibición de la replicación | Planta de té |
| Enmozhi, SK; et al. 2020 (76) | Andrographolide | Interactúa en el sitio de unión de la Mpro | Inhibición de la replicación | Andrographis paniculata |

