










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Medica Herediana
versión impresa ISSN 1018-130Xversión On-line ISSN 1729-214X
Rev Med Hered v.15 n.3 Lima jul./set. 2004
Reporte de un probable caso de Hemoglobina S / Talasemia Beta
Hemoglobin S / Beta Thalassemia. Report of a probable case.
Ramírez Cuentas, John*; Lizama Olaya, Olga**; Martínez La Rosa, Josilú**; Jhong Olivera, Mercy***; Salazar Lindo, Eduardo &.
*Médico Cirujano. Facultad de Medicina Alberto Hurtado de la Universidad Peruana Cayetano Heredia.
** Médico Residente de Pediatría. Universidad Peruana Cayetano Heredia. Hospital Nacional Cayetano Heredia.
*** Médico Residente de Hematología. Universidad Peruana Cayetano Heredia. Hospital Nacional Cayetano Heredia.
&
Médico Pediatra. Hospital Nacional Cayetano Heredia. Profesor de la Facultad de Medicina Alberto Hurtado de la Universidad Peruana Cayetano Heredia.
SUMMARY
We report the case of a patient with Hemoglobin S / Thalassemia. It is feasible to recognize this infrequent disease by its clinical presentation and the aid of clinical laboratory . On this patient, the diagnosis was established based on the clinical findings, hematological evaluation (with careful observation of the red cell morphology and reticulocyte count) and electrophoretic analysis of hemoglobin. We discusse the physiopathology, clinical manifestations, treatment and alternative of prevention of this disease. (Rev Med Hered 2004;15:173-178).
KEY WORDS: Hemoglobin S, thalassemia, anemia.
INTRODUCCIÓN
La Hemoglobina S (Hb S) / Talasemia beta es una condición en la que coexisten una alteración estructural de la cadena beta (Hb S) y otra anomalía cuantitativa de la misma (talasemia beta). El cuadro clínico es variable, pudiendo presentar hallazgos asociados a talasemia (esplenomegalia e ictericia) y drepanocitosis (crisis dolorosas) cuya severidad dependerá de las cantidades porcentuales de Hb S, Hb A y Hb F que se tenga. Se presenta un caso de la Hemoglobina S/ Talasemia, enfermedad que es bastante frecuente en Africa y en los países mediterráneos pero inusual en el Perú (1). No obstante, es importante reconocer dicha enfermedad por las características de su presentación clínica y por las estrategias de diagnóstico y prevención que existen actualmente.
Caso clínico
En el mes de julio del año 2003, se hospitalizó en el Departamento de Pediatría del Hospital Nacional Cayetano Heredia (Lima-Perú), una paciente mujer de seis años de edad, natural y procedente de Lima, sin antecedente de viajes fuera del lugar de procedencia.
La paciente ingresó con un tiempo de enfermedad de seis días, que se inició con dolor de garganta, razón por la cual acude a un centro de salud, donde le indican amoxicilina 50 mg/kg/día, la cual tomó por 2 días, observando leve mejoría. Cuatro días antes del ingreso, se añade fiebre (38°C, axilar), que cede con Ibuprofeno. Tres días antes del ingreso, se agrega palidez marcada, ictericia progresiva de escleras y mucosas y reaparece pico febril (39°C, axilar). Dos días antes del ingreso, presenta además nauseas y vómitos de tipo bilioso (3 cámaras por día). El día del ingreso, al persistir las manifestaciones clínicas mencionadas, acude nuevamente a centro de salud, donde es evaluada y se le realizan exámenes auxiliares, evidenciándose un hematocrito de 15%, razón por la cual es derivada al Hospital Nacional Cayetano Heredia.
Antecedentes prenatales y natales: Gestación sin complicaciones. Edad materna: 35 años. La paciente es la tercera hija de cuatro hermanos. Todos ellos nacieron a término y fueron producto de partos eutócicos. La madre no tuvo abortos. La paciente nació con un peso de 3500 gramos, sin complicaciones y con indicación de alta a las 48 horas. La paciente recibió todas sus vacunas y su desarrollo psicomotor fue adecuado.
Antecedentes patológicos: Hace 2 años estuvo internada por 2 días en el Hospital del Niño con diagnóstico de "Hepatitis". Desde el primer año de vida hasta la actualidad, ha presentado 2 episodios de ictericia que se autolimitaron. No ha presentado otras patologías. No tuvo cirugías previas. Contacto TBC(-), brucelosis(-), tifoidea (-), bartonelosis (-), historia de reacción adversa a medicamentos (-).
Antecedentes familiares: Padre, de 36 años de edad, sano, contador público. Madre, de 35 años de edad, sana, ocupación: su casa. Ambos nacidos en Lima. Tiene tres hermanos (de padre y madre): todos sanos. Tía materna sufre de anemia crónica, desconociéndose su causa. Resto de familiares no presentan ningún antecedente de importancia.
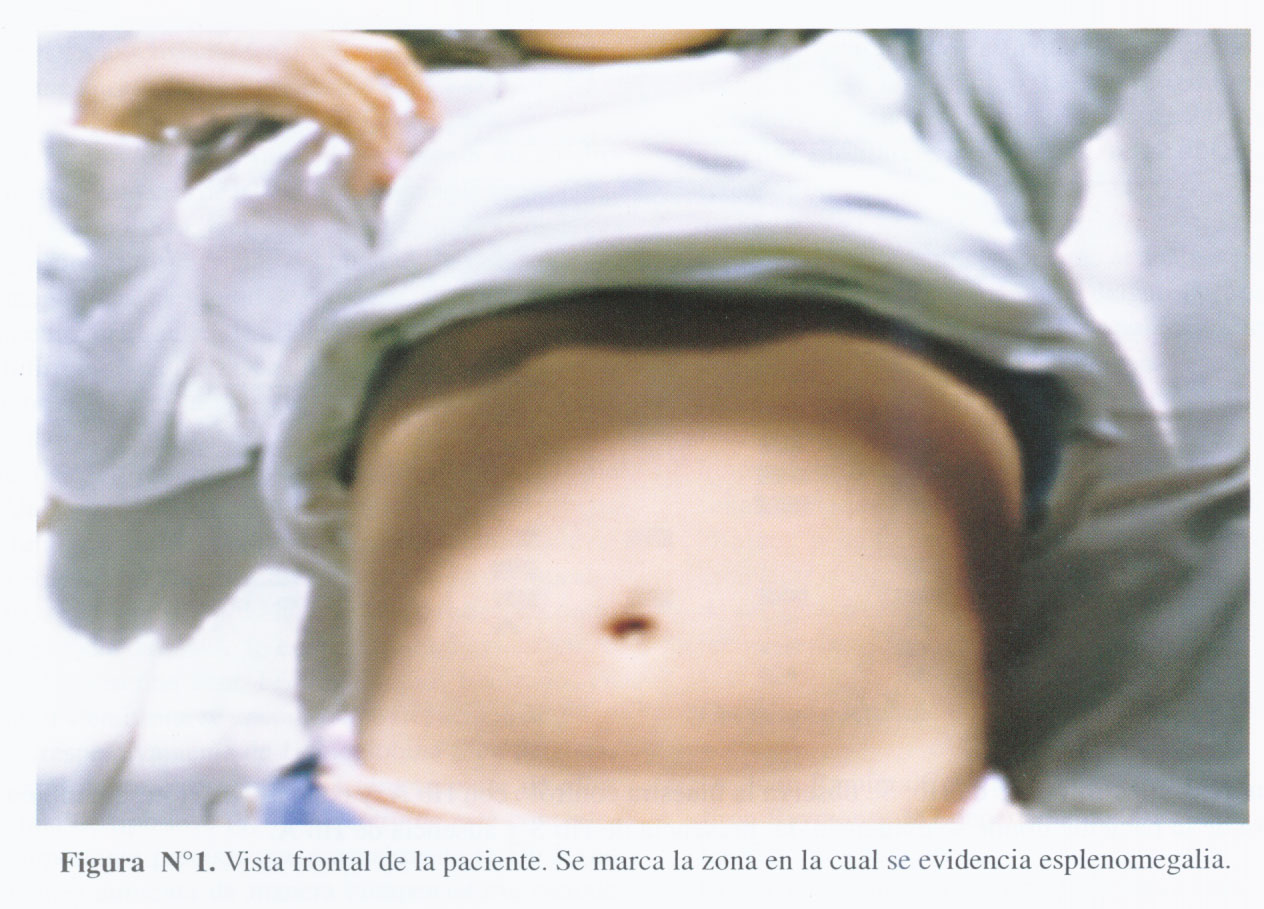
Examen físico: Paciente en aparente regular estado general, hidratada, sin alteración en las funciones vitales ni en los índices peso para edad, talla para la edad y peso para la talla. Hemodinámicamente estable, afebril. Despierta, conectada con el medio. Presentaba marcada ictericia de piel y escleras. Además, en abdomen se evidenció hepatomegalia (tamaño: 11 cm) y esplenomegalia (se palpaba punta de bazo a 2 cm debajo de reborde costal izquierdo línea medioclavicular (Figura N°1). Resto del examen físico: sin alteraciones.
Exámenes auxiliares: Hematocrito de 23%, VCM: 72.5fL; HMC: 24.3pg/célula; CHMC: 33.6g/dL; leucocitos: 8700 / mm3 (2, 78, 1, 0, 2, 17) y plaquetas: 259000/mm3. Recuento de reticulocitos: 17% y prueba de Coombs directa negativa. Se evidenció hiperbilirubinemia (3.34mg/dL) a predominio indirecto (1.80mg/dL). Fosfatasa alcalina: 240UI/L; TGO: 54UI/L; TGP: 16UI/L; DHL: 1981UI/L. Aglutinaciones y examen de gota gruesa negativos. No alteraciones en el examen de orina y urocultivo negativo.
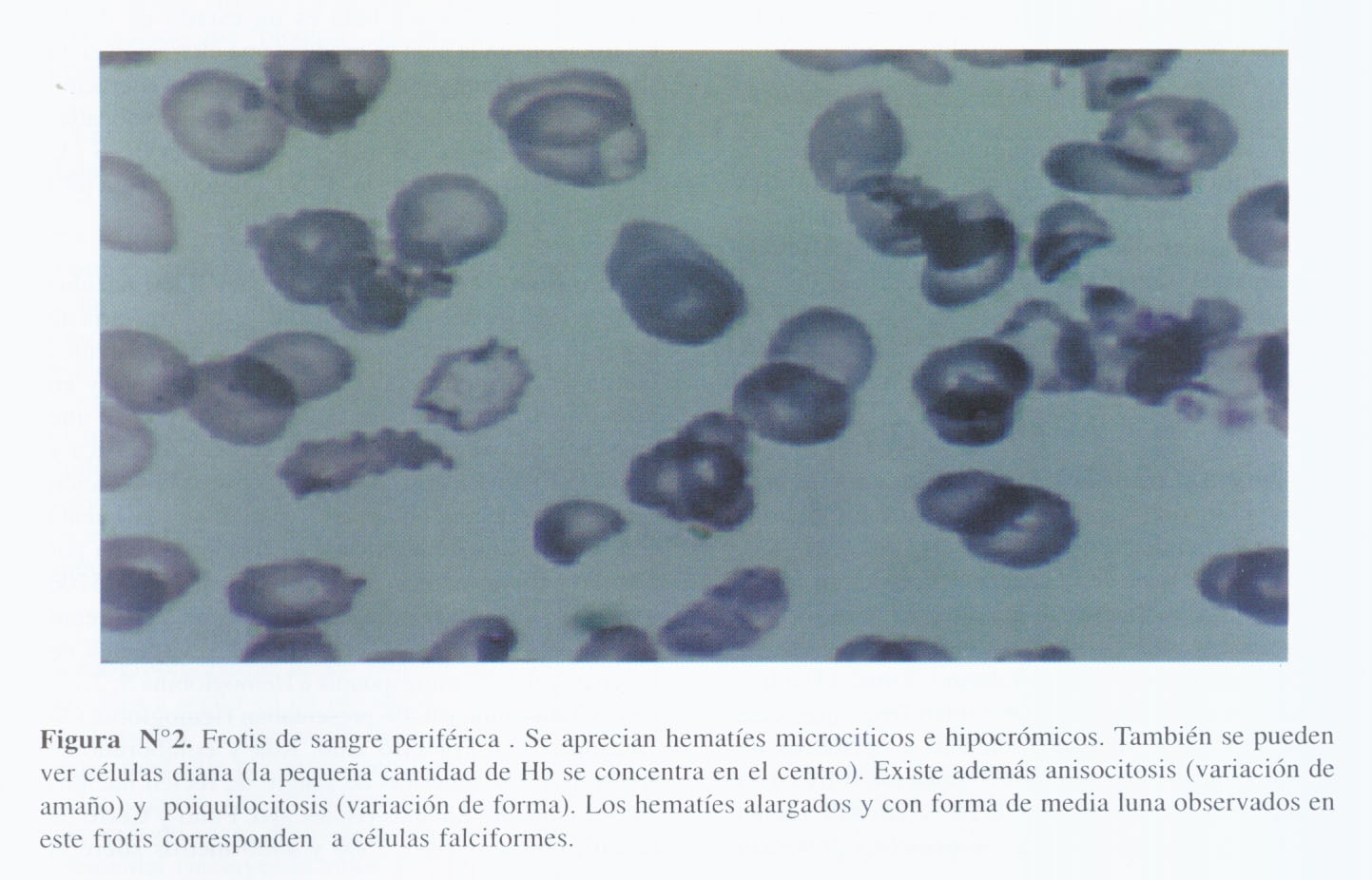
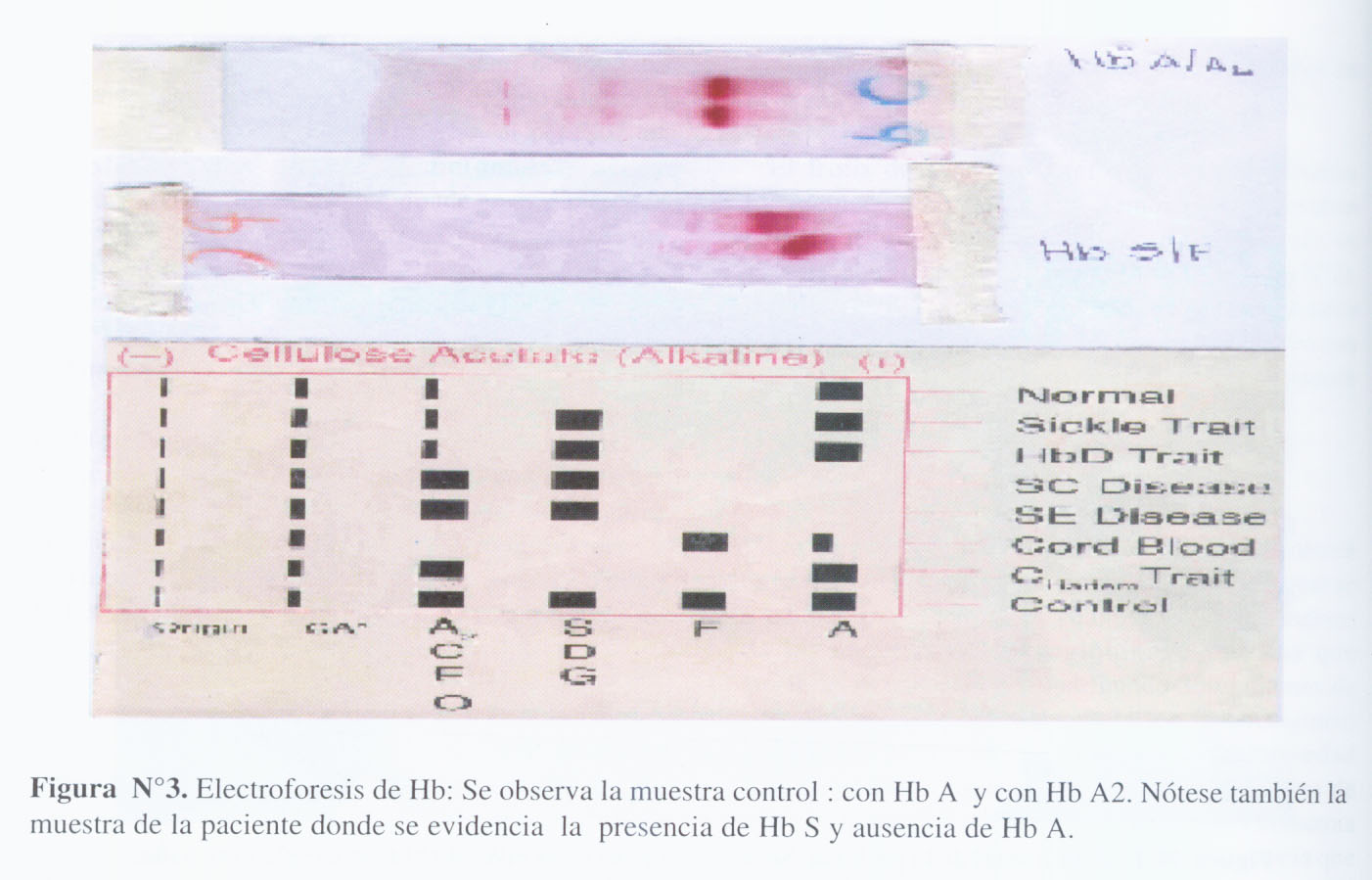
El frotis de sangre periférica mostraba hematíes microcíticos e hipocrómicos. También se observaban células diana y células falciformes. Además se evidenciaba anisocitosis y poiquilocitosis (Figura N°2). La electroforesis de Hb no mostró la banda correspondiente a la Hb A y solamente se observó una banda intermedia localizada a nivel de la que corresponde a la Hb S (Figura N°3).
DISCUSIÓN
Las hemoglobinopatías son un grupo heterogéneo de enfermedades heredadas autosómicamente que se producen por alteraciones cuantitativas o cualitativas de las cadenas de hemoglobina. Se estima que anualmente nacen en todo el mundo 200 millones de personas portadoras de esta alteración (heterocigotos) y 300 mil personas que padecen la enfermedad (homocigotos). Clínicamente, las más importantes de todas las hemoglobinopatías son la talasemia y la anemia de células falciformes; siendo de mayor incidencia que otras enfermedades congénitas frecuentes y graves como el hipotiroidismo congénito y la fenilcetonuria (1,2).
El término "talasemia" proviene del vocablo griego: "thalassa": Mar y "aima": Sangre y hace referencia a la elevada frecuencia con que se observa esta enfermedad entre los habitantes de la cuenca marítima que rodea el Mar Mediterráneo. Bajo el nombre de talasemia, se incluye un grupo muy heterogéneo de alteraciones congénitas cuya característica común es un defecto en la síntesis de una o varias cadenas de globina.
Cada tipo de talasemia recibe el nombre de la cadena que deja de sintetizarse: alfa talasemia (falta la cadena alfa) y beta talasemia (falta la cadena beta) (2, 3). En el caso de alfa talasemia, se pueden tener 4 situaciones clínicas: hidropesía fetal (si ocurre deleción que afecta a los cuatro genes de globina), enfermedad por hemoglobina H (deleción de tres genes), rasgo alfa talasémico (deleción de dos genes) y alfa talasemia silente (deleción de un gen). En lo referente a beta talasemia, existe un déficit de síntesis por mutación puntual en las cadenas beta. El trastorno en la producción de la cadena beta de globina ocasiona el descenso o ausencia de Hb A (2 alfa 2 beta) con aumento de Hb A2 (2 alfa 2 delta) y Hb F (2 alfa 2 gamma). Se tienen 3 situaciones clínicas: beta talasemia menor (con presencia de un gen normal en los heterocigotos), beta talasemia mayor o Anemia de Cooley (son homocigotos para los genes de beta talasemia) y beta talasemia intermedia (que incluye variantes leves del estado homocigoto y algunas formas graves de los heterocigotos) En las talasemias el frotis de sangre periférica muestra usualmente microcitosis e hipocromía.
Por otro lado, la anemia drepanocítica, hemoglobinopatía S o enfermedad por células falciformes es una patología causada por mutación puntual que hace que el ácido glutámico sea sustituído por valina en la sexta posición de la cadena beta globina. La clínica de la drepanocitosis incluye palidez de mucosas, fatiga y disminución de tolerancia al ejercicio. Suele haber ictericia e hiperbilirrubinemia a predomino indirecto.
Se presentan también las crisis vasooclusivas (por obstrucción de vasos terminales por las células falciformes): dolor abdominal, úlceras en las piernas, priapismo, necrosis de la cabeza del fémur, síndrome del tórax agudo (fiebre, dolor toráxico e infiltrados pulmonares), déficit visual, convulsiones, déficit neurológico grave e incluso coma. Dichas manifestaciones clínicas se presentan frecuentemente cuando el paciente está sometido a episodios de deshidratación, infección, desoxigenación o frío. Los individuos heterocigotos para la Hb S (Hb A/S) (rasgo falciforme) usualmente son asintomáticos y presentan sintomatología cuando se exponen a situaciones de estrés hipóxico (2,3,4).
La Hb S/ Talasemia beta es un estado de doble heterocigoto en la cual se produce una alteración en la síntesis de globina y por otro lado ocurre una alteración estructural en la hemoglobina. De las hemoglobinopatías "doble heterocigoto" la más frecuente es la Hb S / C (132/100000) seguida de la Hb S / Talasemia beta (60/100000) en población negra norteamericana.
Así mismo, se estima que 1 de cada 3200 nacidos vivos presentan hemoglobinopatías. Esta cifra varía de acuerdo a la ubicación geográfica y antecedentes familiares, siendo bastante frecuente en Africa y en países mediterráneos. En América, los países que presentan una alta frecuencia son Cuba, Costa Rica y Brasil (5,6). En Perú, el conocimiento sobre la situación actual de las hemoglobinopatías es limitado. Se cuenta con información de un trabajo realizado en Lima por Castillo y colaboradores (1998) quienes estudiaron 5206 muestras de sangre, encontrando que un 7.2% eran hemoglobinas anormales, con 16 tipos de variantes, de los cuales el 3.2% correspondía a Hemoglobina S; 2.1% tenían Talasemia y 0.4% presentaban Hemoglobina S/ Talasemia (7). Por otro lado, Roa y colaboradores analizaron 234 muestras de sangre de recién nacidos de tres ciudades andinas (Huancayo, Puno y Cerro de Pasco), ubicadas entre 3500 y 4400 metros sobre el nivel del mar. En dicha evaluación, no se detectó ninguna variante anormal de hemoglobina (8).
La Hemoglobina S/Talasemia presenta un cuadro clínico y exámenes de laboratorio propios de una anemia de tipo hemolítica. Este es el caso de nuestra paciente, quien presentó ictericia, disminución en el valor de hemoglobina, elevación del número de reticulocitos y aumento en el nivel de la deshidrogenasa láctica y de la bilirrubina indirecta. Las pruebas de laboratorio más importantes para establecer el diagnóstico son: frotis de sangre periférica, test de falciformismo, cuantificación de los niveles de fierro (aspirado de médula ósea) y electroforesis de Hb. Esta última prueba es fundamental para poder determinar la presencia de Hb S y la presencia o ausencia de Hb A. En nuestro caso, se encontró la presencia de Hb S y no se encontró la presencia de Hb A, lo cual apoya el diagnóstico de Hb S/ Talasemia beta. En dos estudios realizados en Huaral y en Lima, se encontró una frecuencia de 3% y 10% respectivamente de Hemoglobina S en personas de raza negra, presentando la tercera parte de ellos anemia de tipo leve (9, 10). Dentro de este contexto, es importante precisar que normalmente la Hb F (no mutante) aumenta de manera compensatoria cuando hay disminución de la Hb A; la cuantía de este incremento es responsable en gran parte de aminorar las manifestaciones clínicas de los pacientes con Hb SS (y de los Hb A/S en situaciones de situaciones de estrés) dado que "bloquea" la cicatrización de la Hb anormal.
En cuanto al tratamiento de la Hemoglobina S/Talasemia beta, las alternativas de manejo incluyen el uso de transfusiones (utilizando quelantes de fierro, por el riesgo de hemocromatosis) y ácido fólico. También es importante una hidratación adecuada, así como el uso de sintomáticos y antibióticos (en el caso de infección concomitante). Así mismo, la esplenectomía y el transplante alogénico de médula ósea constituyen posibilidades terapéuticas. En nuestro caso, el tratamiento se basó en la hidratación, el uso de sintomáticos y ácido fólico, evidenciándose franca mejoría. Por la evolución clínica de la paciente y por su estado hemodinámico, no fue necesario el uso de transfusiones.
La prevención y el diagnóstico prenatal de las hemoglobinopatías deben considerarse alternativas eficaces a seguir en las estrategias de manejo de esta enfermedad. La prevención debe procurarse en primer lugar mediante el consejo genético de los portadores heterocigotos. Los portadores pueden identificarse la mayoría de veces mediante un examen hematológico elemental (incluyendo valor de Hb, frotis de sangre periférica y sideremia) así como la realización de una electroforesis convencional de hemoglobina. Otra alternativa diagnóstica incluye la síntesis in vitro de las cadenas globínicas con sangre fetal obtenida por fetoscopía.
Finalmente debe precisarse, que el análisis del ADN tiene un valor primordial en el diagnóstico prenatal. Mediante la reacción en cadena de la polimerasa se puede amplificar el ADN obtenido por biopsia de vellosidades placentarias o por amniocentesis, para luego detectar fácilmente la alteración que dio origen a la hemoglobinopatía (2,5,11). Queremos finalizar señalando, que en este caso se emplearon criterios clínicos y métodos accesibles de laboratorio para establecer el diagnóstico de una enfermedad que a pesar de ser poco frecuente en nuestro medio, es importante reconocerla.
Agradecimientos:
Queremos agradecer a quienes contribuyeron con su valiosa ayuda en la realización del presente trabajo:
- Dr. Wilson Ruiz Gil: Médico Hematólogo del Hospital Nacional Cayetano Heredia. Docente de la Universidad Peruana Cayetano Heredia.
- Dra. Elsa Chea Woo: Médico Pediatra del Hospital Nacional Cayetano Heredia. Docente de la Universidad Peruana Cayetano Heredia.
- Dr. Eduardo Negrón Saavedra: Médico Pediatra del Hospital Nacional Cayetano Heredia. Docente de la Universidad Peruana Cayetano Heredia.
REFERENCIAS BIBLIOGRÁFICAS
1. Haj A, Laradi S, Miled A, Omar G, Ben J, Perrin P. Clinical and molecular aspects of haemoglobinopathies in Tunisia. Clin Chim Acta 2004; 340:127-137. [ Links ]
2. Beutler E. Alteraciones de la Hemoglobina : Harrison: Principios de Medicina Interna. Kurt I, Jean W, Dan L, Anthony F, Eugene B, Stephen H. Editorial McGraw - Hill - Interamericana, Madrid 1998,pp:737-745. [ Links ]
3. Shoen F. Enfermedades de los hematíes y trastornos hemorrágicos. En: Patología Estructural y Funcional. Frederick S. Editorial McGraw-Hill-Interamericana, Madrid 1995, pp:645-694. [ Links ]
4. Gonzales G, Ruiz W. Hemoglobinopatías y la exposición aguda a la altura. Acta Andin 1997;6:3-10. [ Links ]
5. Orlando G, Naoum P, Siqueira F, Bonini C. Diagnóstico laboratorial de hemoglobinopatías en diferentes poblaciones. Rev Bras Hematol Hemoter 2000; 22:111-121. [ Links ]
6. Martínez G, Hernández A, Corral L, Muniz A, Hernández A. Biología molecular en hemoglobinopatías y en hemopatías malignas. Rev Cubana Hematol Inmunol Hemoter 1996; 12:81-85. [ Links ]
7. Castillo J, Hazán E, Márquez M. Las Hemoglobinopatías en el Perú. Rev Med Inst Peru Segur Soc 1998; 7:7-17. [ Links ]
8. Roa D, Aguinaga M, Ruiz W, Ulloa V, Turner E. Búsqueda de Hemoglobinas anormales en los recién nacidos en las grandes alturas. Rev Med Hered 1997; 8:87-91. [ Links ]
9. Román Z. Prevalencia de Hemoglobina S en la población de raza negra de Aucallama – Huaral Tesis de Bachiller. Universidad Nacional Mayor de San Marcos. Lima, Perú. 2000. 28pp. [ Links ]
10. Piaggio F, Ruiz O, Tuesta J, Valdivia G. Estudio de hemoglobinas anormales en personas de raza negra en Lima Metropolitana. Rev Med Peru 1995; 67:50-53. [ Links ]
11. Old JM. Hematological applications: the haemoglobinopathies. Methods Mol Med 2004; 92:203-219. [ Links ]
Correspondencia:
Dr. John Ramírez Cuentas.
Jirón Raymondi 387 departamento 201
La Victoria. Lima, Perú.
Correo electrónico: locojohn22@hotmail.com


{kind=link}
{kind=link}
{kind=link}