











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkPRESENTACIÓN DEL CASO
Dra. Carla Alvarez (Residente de 3er año de Medicina Interna)
Mujer de 49 años con un tiempo de enfermedad de 12 días, caracterizado por dolor abdominal tipo cólico, intensidad 4/10, difuso, no irradiado, asociado a tenesmo rectal, polaquiuria y disuria. Además, notó discreta debilidad en miembros inferiores. Por la persistencia de las molestias acudió a un centro médico donde le realizan una TEM abdominal sin contraste y le refieren que presenta una tumoración retroperitoneal y le aconsejan acudir a un centro especializado y fue dada de alta. Presentó mayor debilidad en miembros inferiores, a predominio izquierdo, por lo que tenía que caminar con ayuda; además, se agregó retención urinaria y fecal. Acudió a otro centro médico donde le colocaron sonda urinaria por globo vesical y recomiendan hospitalización. Dos días antes del ingreso la debilidad en miembros inferiores progresa y ya no podía deambular. Acudió a nuestro hospital donde fue admitida.
Funciones biológicas: Apetito conservado; sed conservada; sueño alterado, refiere dificultad para conciliar el sueño; peso: pérdida de 5 kg en 10 días; sudor sin alteración; orina, disuria; deposiciones disminuido en frecuencia, refiere haber realizado ultimas deposiciones con laxante y enema. Estado de ánimo, tristeza.
Antecedentes: Gineco-obstétricos: G1 P1001. FUR 31/10/20. No usaba métodos anticonceptivos. La paciente había tenido herpes zoster dorsal hace 3 años, sin secuelas. Negaba otras enfermedades, hospitalizaciones y cirugías. Negaba alergias medicamentosas y no consumía fármacos. En los antecedentes familiares tenía una prima hermana con lupus eritematoso sistémico.
La revisión anamnésica de sistemas y aparatos fue no contributoria.
El examen físico mostró PA: 100/70 mm Hg, FC: 90 latidos/minuto, FR: 18 respiraciones/minuto y T oral 36,6°C. Estaba despierta y orientada en las tres esferas. La piel era normal, no había edema ni adenopatías. El examen de cabeza y cuello, cardiovascular y tórax eran normales. El abdomen era blando, no doloroso al momento de la evaluación y era portadora de sonda vesical. El examen neurológico mostraba escala de Glasgow 15, pupilas isocóricas y foto reactivas, pares craneales conservados. La evaluación de la fuerza reveló tono flácido y debilidad severa en ambas piernas, asimétrica, 3/5 en pierna derecha y 0/5 en izquierda. Los reflejos osteotendinosos estaban aumentados, con clonus y signo de Babinski bilateral en miembros inferiores. Había alteración en la sensibilidad en dichos miembros y nivel sensitivo en D5. La fuerza, sensibilidad y reflejos en miembros superiores estaban normales.
Los análisis de laboratorio mostraron hemoglobina 13,2 g/dl, leucocitos 5060/mm3 con un recuento diferencial normal excepto linfopenia de 10%, plaquetas 353 000 / mm3. Glucosa, creatinina, electrolitos, calcio y ácido úrico séricos fueron normales. El perfil de coagulación no mostró anormalidades y tampoco el perfil hepático excepto una leve hipoalbuminemia (3 gr/dl). La proteína C reactiva cualitativa fue negativa y deshidrogenasa láctica 142 U/l (VN: 140-280). Los test de ELISA para VIH, HTLV1, HBsAg y hepatitis C resultaron negativos; beta-2 microglobulina sérica 1,2 mg/l (VN 0,7 - 2,1) y anticuerpos antinucleares (ANA) con patrón moteado 1:160. La radiografía de tórax no mostró alteraciones.
La tomografía abdominal sin contraste mostró una tumoración sólida de probable origen neoformativo en espacio pararrenal anterior izquierdo (retroperitoneo) parcialmente valorada por falta de la fase contrastada, asociada a adenopatías retroperitoneales ipsilaterales (Figura 1). La resonancia magnética nuclear (RMN) mostró alteración de la señal en la medula espinal de la región cervical y dorsal de C5 a T10, luego de la administración de contraste se observa áreas de realce en esas regiones. Los cuerpos vertebrales sin evidencia de lesiones expansivas ni colapsos (Figura 2). Además, la RMN encefálica evidenció lesiones desmielinizantes en la región posterior del tronco del cuerpo calloso, en la región posterior de los cíngulos a predominio izquierdo y en la región posterior al cuerno occipital del ventrículo lateral izquierdo (Figura 3).
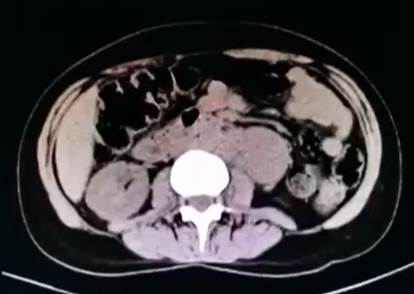
Figura 1 Tomografía abdominal sin contraste. En espacio pararrenal anterior izquierdo y por delante del psoas iliaco, se visualiza tumoración solida de 5,9 x 3,7 cm de diámetro transverso y hasta 7,3 cm de longitud, tiene probable origen neoformativo y asocia adenopatías retroperitoneales ipsilaterales de hasta 1,5 cm de eje corto.
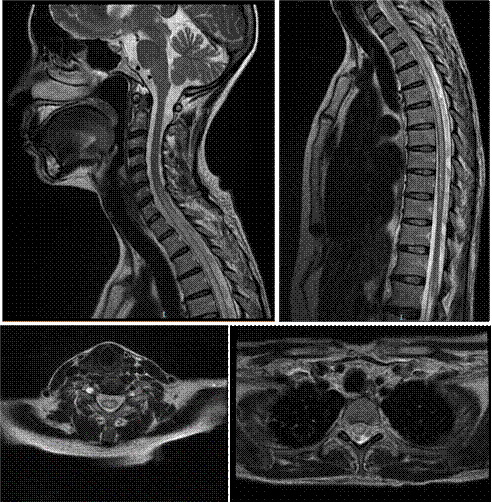
Figura 2 Resonancia magnética nuclear de columna total en múltiples secuencias. Se muestran secuencias en T2 con alteración de la señal en la medula espinal de la región cervical y dorsal de C5 a T10. Luego de la administración de contraste en se observó realce de estas lesiones. Estas alteraciones son compatibles con lesiones desmielinizantes. Los cortes axiales muestran que la lesión es central.
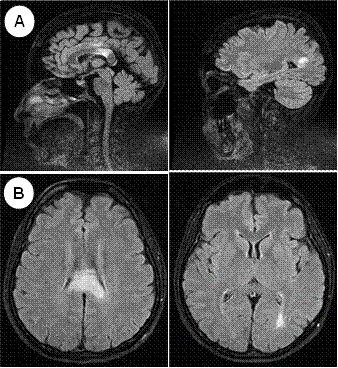
Figura 3 A: RMN de encéfalo sagital - FLAIR: Hiperintensidad en cuerpo calloso (esplenio, cíngulo), cuerno posterior del ventrículo lateral izquierdo. B: RMN de encéfalo axial - FLAIR: Hiperintensidad en cuerpo calloso, cuerno posterior del ventrículo lateral izquierdo.
El estudio de líquido cefalorraquídeo, mostró presión de apertura de 16 cm H2O, incoloro, transparente, 2 leucocitos/mm3, 6 hematíes/mm3, proteínas y glucosa normales, panel meningitis/encefalitis por Film array: Bacterias: E. coli, H. influenza tipo B, L. monocytogenes, N. meningitidis, S. agalactiae, S. pneumoniae: negativos. Virus: CMV, Enterovirus, Herpes simplex virus 1 y 2, Human herpes virus 6, Parechovirus, Varicella zoster virus: negativos. Hongos: C. neoformans/gattii, negativo. No se detectaron bandas oligoclonales de IgM e IgG en LCR.
DISCUSIÓN
Dr. Sergio Vásquez Kunze (Medicina Interna)
Esta paciente tiene una historia clínica caracterizada por dos síntomas cardinales: dolor abdominal y paraparesia. El dolor abdominal es transitorio pero la paraparesia es progresiva y se convierte en el principal problema para la paciente. Cuando se presenta un problema neurológico lo primero es delimitar su topografía y luego documentarla con una imagen si es apropiado. Luego de eso a partir de los hallazgos en las imágenes y la historia clínica plantear las principales posibilidades diagnósticas. Los síntomas y signos neurológicos en esta paciente como la paraparesia, la hiperreflexia, signo de Babinski bilateral, compromiso de esfínteres y nivel sensitivo, identifican claramente una lesión en médula espinal, es decir un síndrome medular. Esta lesión puede ser intrínseca como una mielitis transversa o extrínseca a la medula como una lesión vertebral y compresión de ésta. La mejor imagen para caracterizar un síndrome medular es una RMN contrastada. Esta se solicita de acuerdo con el nivel sensitivo encontrado y con visualización adicional de los demás segmentos pues las lesiones pueden ser multifocales. En la paciente la RMN de médula espinal muestra una extensa lesión hiperintensa en secuencia T2 que capta contraste desde C5 a T10. Que sea hiperintensa en T2 y se realce con contraste es compatible con una lesión inflamatoria. Una lesión medular inflamatoria no compresiva subaguda como ésta es compatible con una mielitis transversa (MT). Cuando una lesión medular inflamatoria tiene una extensión de tres o más segmentos contiguos se denomina mielitis transversa longitudinalmente extensa (MTLE), en contraposición a la mielitis transversa más común de una menor longitud. La MTLE es una clave importante para hacer más específico nuestro diagnóstico diferencial 1 (tabla 1).
Tabla 1 Diagnostico diferencial de mielitis transversa longitudinal extensa
| - Desordenes del espectro de la neuromielitis óptica |
| - Anti MOG |
| - Enfermedades autoinmunes (LES, ES, SAF) |
| - Sarcoidosis |
| - Esclerosis múltiple |
| - Estados infecciosos y post infecciosos (HTLV 1, HIV, CMV, EBV, TBC, sífilis, esquistosomiasis) |
| - Neoplasias |
| - Vasculares (isquemia, fistulas arteriovenosas) |
| - Metabólicas (déficit Vitamina B12, cobre) |
(ES: Esclerosis sistémica; SAF: Síndrome Antifosfolípido; CMV: Citomegalovirus; EBV: Epstein Barr virus; TBC: Tuberculosis)
El segundo problema es el dolor abdominal, que es transitorio y probablemente debido a la misma mielitis transversa que puede caracterizarse con un dolor lancinante, inicialmente en cinturón. Esto llevó a realizar una TEM abdominal que reveló una tumoración retroperitoneal que impresionan masas ganglionares. El diagnóstico diferencial puede ser amplio e incluyen neoplasias como linfoma o sarcomas. Por la localización esta lesión de masa requerirá una biopsia laparoscópica o por radiología intervencionista.
El diagnóstico sindrómico de MTLE en este paciente nos servirá para discutir nuestro diagnóstico diferencial y plan diagnóstico final.
Mielitis transversa longitudinalmente extensa (MTLE)
Enfermedades inflamatorias sistémicas: Enfermedades autoinmunes como lupus eritematoso sistémico (LES), Sjögren o sarcoidosis pueden debutar con MT 2-4. Se debe buscar otros síntomas reumatológicos o de sarcoidosis para mejorar nuestra probabilidad diagnóstica. Indagar sobre antecedentes familiares puede ser clave. Según el juicio clínico pueden ser útiles: PCR, VSG, ANA, anti Ro/SSA, anti La/SSB), panel antifosfolipídico, perfil ENA, ANCA y complemento. Revisar la radiografía de tórax para adenopatías hiliares puede sugerir sarcoidosis. Esta paciente tiene una prima hermana con LES y un ANA positivo. Aunque no tiene otros síntomas de LES y el ANA es inespecífico, se deberían completar los exámenes arriba descritos para descartar esta posibilidad.
Infecciones: Varias infecciones pueden ser causa, aunque raramente, de una MT. Virus, bacterias como Brucella o Mycobacterium tuberculosis5 y otras bacterias atípicas, parásitos y hongos, pueden ser sospechados a partir de claves en la historia clínica como fiebre, rash, adenopatías, cefalea o meningismo. Nuestra paciente no presenta estos síntomas o signos y no tiene claves epidemiológicas para su sospecha. Los exámenes para dichas infecciones en LCR fueron negativos, lo cual aleja esta posibilidad.
Deficiencias nutricionales: Deficiencias de vitamina B12, cobre y vitamina E, pueden causar mielopatía 6. Aunque la historia clínica para estas deficiencias frecuentemente revela malnutrición asociada a malabsorción, cirugía bariátrica o aumento de consumo de zinc (que causa deficiencia de cobre), en otras no se encuentra un factor predisponente claro y el dosaje de estas vitaminas y oligoelementos puede ser de utilidad. La deficiencia de cobre produce una mielopatía en región dorsal con ataxia sensitiva, que puede ser indistinguible del déficit combinado subagudo de la deficiencia de vitamina B12. Nuestra paciente tiene una lesión medular central lo cual hace improbable estas deficiencias como causa de la mielopatía.
Enfermedades inflamatorias desmielinizantes del SNC: En un paciente con MT es mandatorio realizar una RMN de encéfalo con gadolinio para diagnosticar lesiones desmielinizantes asintomáticas asociadas. Las lesiones multifocales en médula espinal y encéfalo hacen sospechar una enfermedad desmielinizante del SNC en nuestra paciente. Estas son: la esclerosis múltiple (EM) la encefalomielitis diseminada aguda (ADEM), el espectro de enfermedad de la neuromielitis óptica (NMOSD) y la enfermedad por anticuerpos anti-MOG. En la EM la lesión medular es de menor extensión que la presentada por la paciente. Las lesiones en encéfalo tampoco son típicas de EM 7. De estas las que presentan una lesión extensa en la medula espinal (MTLE) son característicamente la NMOSD y enfermedad por anti MOG (glicoproteína de la mielina del oligodendrocito). Esta paciente tiene un cuadro clínico e imágenes compatibles con NMOSD.
La NMOSD se caracteriza por presentar ataques agudos y recurrentes de MT o neuritis óptica 8. También pueden presentar síntomas encefálicos como nauseas intratables, hipo, encefalopatía o somnolencia. La lesión medular es frecuentemente una MTLE como en nuestra paciente. Las lesiones encefálicas pueden localizarse en el área postrema del bulbo (causando náuseas, vómitos e hipo) (9, 10), tronco cerebral, diencéfalo (causando sueño excesivo diurno, SIADH) 11 y lesiones peri ependimarias extensas en cuerpo calloso. La hiperintensidad extensa en el cuerpo calloso y la MTLE es muy sugerente de NMSOD en esta paciente. La etiología de esta enfermedad es autoinmune; los anticuerpos identificados son los dirigidos contra la acuaporina 4 (anticuerpos anti-AQP4) 12. ANA y anti SSA/SSB son frecuentes en la NMOSD, los cuales también presenta esta paciente. La enfermedad por anticuerpos anti-MOG comparte algunas similitudes con NMOSD, aunque se asocia más frecuentemente con síntomas parecidos al ADEM y las lesiones medulares suelen ser múltiples 13, las cuales no presenta nuestra paciente.
Enfermedades paraneoplásicas: Es importante discutir estas enfermedades pues esta paciente tiene una lesión de apariencia tumoral abdominal ya descrita. Primero, las mielopatías paraneoplásicas se presentan frecuentemente como MTLE. La neoplasia más frecuente es el cáncer de pulmón de células pequeñas. Estas mielopatías se producen por ataque a la medula espinal por anticuerpos, más comúnmente anti-Hu y anti CRMP5 14. La asociación de NMOSD con anticuerpos para AQP4 y neoplasias esta descrita en serie de casos 15. Más comúnmente se asocian a cáncer de mama y de pulmón. Al tener esta paciente una presentación compatible con NMOSD y una tumoración abdominal, se debe sospechar fuertemente un NMOSD paraneoplásico.
Idiopática: Frecuentemente no se encuentra alguna condición asociada 16. Por lo descrito anteriormente es improbable que este paciente no tenga una causa demostrable.
En esta paciente, el plan de trabajo adicional incluiría dosaje sérico de anticuerpos anti AQP4, anti-MOG y una laparoscopía diagnóstica para biopsia de la tumoración abdominal. Debe empezar pulsos de metilprednisolona y plasmaféresis para la MT.
EVOLUCIÓN
Se realizaron exámenes de anti-AQP4 IgG sérico cuyo resultado fue > 10 U/l (VN: < 1 = negativo). Los anticuerpos contra la mielina oligodendrocito glicoproteína (MOG) fueron negativos. La evaluación por oftalmología no encontró neuritis óptica.
El paciente respondió sub óptimamente a pulsos de metilprednisolona 1 gr cada 24 h por 5 días, por lo cual posteriormente se administró 7 ciclos de plasmaféresis, con poca mejoría.
La paciente fue intervenida con una mini laparotomía exploratoria y se realizó la extracción de un ganglio retroperitoneal que mostró proliferación linfoide atípica con células sternbergoides sugestivas de proceso linfoproliferativo (figura 4). La inmunohistoquímica fue positiva para CD30, y PAX5, Ki67 40% y CD20, CD3, CD15 y EBV negativos; compatible con linfoma de Hodgkin clásico, variedad esclerosis nodular. El estudio de citometría de flujo en el LCR no detectó infiltración de células neoplásicas de linfoma de Hodgkin.
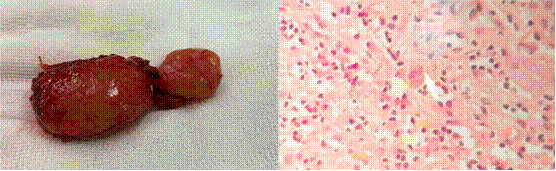
Figura 4 Ganglio retroperitoneal y vista histológica: Proliferación linfoide atípica con células sternbergoides sugestivas de proceso linfoproliferativo.
La fuerza muscular de las extremidades inferiores aumentó a 2/5 en miembro inferior izquierdo y 4/5 en miembro inferior derecho. Se inició quimioterapia con esquema ABVD: Bleomicina 15 mg, doxorrubicina 35 mg, dacarbazina 550 mg y vinblastina 9 mg. Fue dada de alta, continuando controles y quimioterapia por consultorio externo. Dos meses después, solo tenía una leve debilidad en la pierna izquierda (4/5), mientras que la fuerza motora de la pierna derecha era normal. Mejoró notablemente la sensibilidad y no presentaba alteraciones esfinterianas.
Martín Tipismana (Neurología)
Nuestra paciente, mujer de 49 años, presenta un cuadro subagudo de dolor abdominal y paraparesia inicialmente asimétrica. La paraparesia progresa, encontrándose hallazgos clínicos de primera neurona motora (paraparesia espástica), compromiso autonómico (globo vesical y estreñimiento) y nivel sensitivo en D5, diagnosticándose clínicamente un síndrome medular. La RMN medular muestra una lesión intramedular cérvico dorsal (C5-T10), con captación de contraste, compatible con una MTLE. La RMN cerebral confirma la presencia de múltiples lesiones periventriculares (cuerpo calloso, ventrículo izquierdo) sin captación de contraste. Ambas lesiones (medulares y cerebrales), compatibles con enfermedad desmielinizante. El estudio de LCR fue normal. Se encontró una masa abdominal (adenopatía) compatible con un Linfoma de Hodgkin clásico en anatomía patológica.
Los diagnósticos diferenciales de la enfermedad desmielinizante fueron descartados (esclerosis múltiple, post infecciosa, toxico-metabólica, autoinmunidad), encontrándose el marcador de anti-AQP4 IgG sérico positivo. Entonces, se confirma por clínica, radiología y laboratorio un cuadro de Desorden del espectro de neuromielitis óptica (NMOSD) 17. En este caso, no es NMOSD idiopático (primario), es secundario (paraneoplásico).
NMOSD, es un grupo de trastornos inflamatorios que afecta más a mujeres, compromete el SNC y se presenta en formas monofásicas y generalmente recurrentes, de desmielinización y daño axonal mediado por el sistema inmunitario que compromete principalmente los nervios ópticos y la médula espinal. Actualmente se reconoce que la presencia del marcador anti AQP4-IgG sérico induce reacción inflamatoria, incluyendo citotoxicidad dependiente del complemento, infiltración celular, citotoxicidad celular dependiente de anticuerpos resultando en daño astrocitario llevando secundariamente a la desmielinización y la injuria neuronal. AQP-4 es una proteína que forma parte de un canal de agua, ubicada principalmente en los pies astrocitarios de la barrera hematoencefálica y en áreas periventriculares. También han sido ubicadas en otros tejidos: células parietales del estómago, tubos colectores, vías respiratorias, glándulas secretoras y musculares 18,19.
La incidencia y prevalencia es de 0,05-0,4 y 0,52-4,4 por 100 000 habitantes, respectivamente 19. En los pacientes con NMOSD, el 70 - 80% tienen positividad para anti-AQP4, y 10 a 40 % de los anti-AQP4 negativos, son positivos para anti-MOG 19,20. En relación a una condición subyacente, se reporta enfermedades autoinmunes en 25% de pacientes con NMOSD y antiAQP4 presentes 19. La presencia de síndrome paraneoplásico asociado a NMOSD es reportado en la literatura con una baja prevalencia y parece ser más frecuente en población adulta mayor. En una serie de casos se reporta una prevalencia de 3,2 % (5/156), siendo los tumores más frecuentes de pulmón y de mama. También están asociados a teratoma ovárico, meningioma, timoma, tumor endocrino 20,21. Estas neoplasias pueden estar presentes antes o después del diagnóstico de NMOSD.
La presentación clínica de nuestra paciente fue la de una paraparesia bilateral, asimétrica que progresa definiéndose en una mielopatía. La forma de presentación típica de NMOSD es con compromiso medular, visual, y con menos frecuencia troncales y afectaciones encefálicas / hipotalámicas (encefalopatía, convulsiones, narcolepsia). En los casos reportados, la afectación medular es la más frecuente, seguida del compromiso óptico y del área postrema 17.
En nuestra paciente se encontraron lesiones tipo MTLE y lesiones en sustancia blanca periventriculares en los estudios de RMN con contraste. Las lesiones medulares fueron de preferencia centrales con captación de contraste. Las lesiones encefálicas no captaron contraste. Los reportes en la literatura no son diferentes a nuestros hallazgos.
En relación al LCR, la paciente tiene una liquido de características normales; la búsqueda de marcadores virales herpéticos, enterovirus, BOC, anti-MOG fueron negativos. Como en otras enfermedades inflamatorias no infecciosas, (ej. Esclerosis Múltiple), la literatura registra discretos incrementos de la celularidad, proteinorraquia y presencia de BOC en menos del 20% (en esclerosis múltiple es más del 80%). Varios reportes realizan búsqueda de otros marcadores paraneoplásicos de afectación neurológica, como CRMP5 y anphiphysin (mielopatías y síndrome de hombre rígido, respectivamente) 20-23.
Inicialmente, nuestra paciente recibió pulsos de MP por 5 días y luego 5 sesiones de plasmaféresis, con discreta mejoría neurológica. Durante su estancia hospitalaria no hubo otro compromiso neurológico y posteriormente recibió tratamiento específico con lo cual se evidencia una recuperación neurológica importante en los tres meses siguientes. El manejo propuesto en la literatura es iniciar con pulsos de MP y si hoy respuesta añadir plasmaféresis. La inmunoglobulina no tiene un mayor impacto en el tratamiento.

