










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. vol.75 no.1 Lima ene. 2014
CASO CLÍNICO
Tumor maligno de tritón pélvico: reporte de un caso
Pelvic triton malignant tumor: case report
Himerón Perfecto Limaylla Vega1,a, Herberth Alfaro Salazar1,b, Edwin Wilder Damián Bello1,c, José Romero Yauri1,d
1 Hospital Nacional Dos de Mayo, Lima, Perú.
a Cirujano General Jefe del Servicio I-4; b Médico Patólogo Asistente; c Médico Residente de Cirugía General; d Médico Residente de Anatomía Patológica.
Resumen
Se presenta el caso de una paciente de 29 años con dolor en la región glútea y miembro inferior derecho, portadora de una enfermedad de Von Recklinghausen y que desarrolló dos tumoraciones pélvicas retroperitoneales de crecimiento rápido. En la laparotomía se encontró las tumoraciones adheridas al hueco obturatriz y alerón del hueso iliaco. Se procedió a exéresis de las tumoraciones, la del lado derecho de mayor tamaño, de evolución rápida y compatible con tumor de Tritón, y la izquierda compatible con neurofibroma.
Palabras clave: Neurofibromatosis, tumor de tritón.
Abstract
We report the case of a 29 year-old woman complaining of pain in right buttock and leg. She suffered of von Recklinghausen disease and developed rapidly growing retroperitoneal pelvic tumors. At laparotomy the tumors were attached to the obturator foramen and the iliac wing. The larger and fast growing right side tumor compatible with malignant triton tumor, and the left sided neurofibroma were removed.
Keywords: Neurofibromatosis, triton tumor.
INTRODUCCIÓN
Los elementos de rabdomiosarcoma dentro de los tumores malignos de la vaina de los nervios periféricos (MPNSTs) fueron descritos por Masson en pacientes con neurofibromatosis (NF-1) (1,2). El nombre de tritón fue usado primero por Woodruff y col (3), basándose en el descubrimiento de que los miembros supernumerarios que contienen elementos neuronales y musculares fueron inducidos a crecer sobre las espaldas de las salamandras tritón por trasplante. Woodruff y col (3) propusieron tres criterios para determinar si una neoplasia podría ser clasificada verdaderamente como un tumor maligno de 'tritón' o no: 1) surge de un nervio periférico, o en un paciente con NF-1 o en un lugar típico para tumores de nervio periférico, o representa una metástasis de tal tumor; 2) muestra las características de crecimiento de las células de Schwann; y, 3) contiene rabdomioblastos que parecen surgir de dentro del cuerpo del tumor de nervio periférico y que no puede atribuirse a una extensión o metástasis de un rabdomiosarcoma extrínseco. Daimaru y col (4) más adelante sugirieron ampliar la definición para incluir lo siguiente: 1) tumores en los pacientes sin NF-1 que son microscópicamente compatibles con un schwannoma maligno y contienen rabdomioblastos focales; y, 2) tumores compuestos predominantemente de diferenciación rabdomioblástica con elementos de la célula de Schwann focal que ocurren dentro de un nervio o en pacientes con NF-1. Hoy en día, generalmente se hace el diagnóstico de tumor maligno de tritón (MTT) según estos criterios, así como con los resultados de la inmunohistoquímica (5). La presencia de inmunorreactividad positiva a la proteína S-100 o Leu-7 indica la diferenciación de la vaina del nervio, mientras que las células de rabdomioblastos tienen una inmunoreactividad positiva para desmina, actina específica de músculo, miosina, vimentina y mioglobulina (3,5,6).
Reportamos este caso en una mujer joven, debido a la rareza del tumor de tritón, sobre todo de localización retroperitoneal y a nivel pélvico, por la conducta agresiva de dicho tumor, gran recidiva local y capacidad de metástasis a distancia, a pesar de tratamiento quirúrgico radical, quimioterapia y radioterapia complementarios, haciendo sombrío su pronóstico, con un promedio de sobrevida de 26% (6).
PRESENTACIÓN DEL CASO
Paciente de sexo femenino de 29 años de edad, con antecedente quirúrgico de cesárea hacía tres años, por pelvis estrecha. Acudió al servicio de cirugía referida del servicio de ginecología, con un tiempo de enfermedad de 6 meses, caracterizado por dolor tipo hincada en la región glútea y miembro inferior derecho, que se inició en el dedo del pie derecho y progresó hacia el glúteo, aumentando en intensidad, con dolor intenso en la deambulación que no cedía a los analgésicos comunes, siendo derivada al servicio de cirugía oncológica.
Al examen físico preferencial, la piel mostraba múltiples manchas café con leche de diferente diámetro, en espalda, antebrazo y miembros inferiores, compatibles con enfermedad de Von Recklinghausen. En la región inguinal derecha se palpó una adenopatía de 1,5 cm, móvil, lisa, no dolorosa a la palpación, y otra en la región inguinal izquierda de 0,5 cm de diámetro, con las mismas características.
En los exámenes auxiliares, el hemograma, perfil de coagulación, glucosa, urea y creatinina no mostraron alteraciones. La ecografía del 23/07/12 informó que en el anexo derecho se observaba el ovario de 29,8 x 27 mm, con folículo de 8 mm, y tumoración de 60 x 56 mm, de bordes bien delimitados, ecogenicidad ligeramente heterogénea, etiología sin determinar. El ovario izquierdo medía 29 x 16 mm, con folículo de 5 mm y tumoración de 36 x 28mm, con las mismas características de la anterior. La conclusión fue tumoración ovárica bilateral, y se sugirió tomografía espiral multicorte (TEM).
La TEM de abdomen-pelvis con contraste demostró tumoración heterogénea sólida en el lado derecho de 57 x 71 mm, tumoración ovoidea sólida de aspecto dependiente del músculo glúteo mediano, de 48 x 28 mm. Se sugirió ampliar el estudio con resonancia magnética (RM) (figura 1) y descartar sarcoma y/o biopsia. Se evaluó conjuntamente con el servicio de neurocirugía, ginecología oncológica y cirugía cardiovascular, por estar las tumoraciones ubicadas en la pelvis menor y retro-peritoneo.
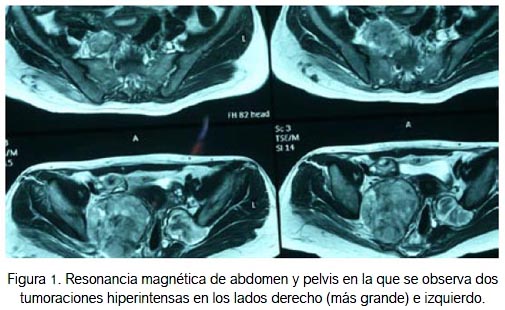
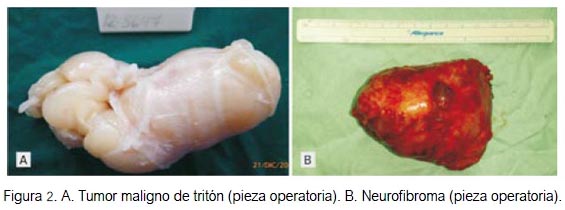
La paciente ingresó a sala de operaciones para una laparotomía exploratoria y posibilidad de exéresis de los tumores retroperitoneales, con el presunto diagnóstico de neurofibromas pélvicos, en una paciente con enfermedad de neurofibromatosis. Se halló una tumoración retroperitoneal pélvica derecha de 9 x 6 cm, de consistencia dura, localizada a nivel de las raíces nerviosas L5-S1 y adherida al agujero obturatriz, al alerón del hueso ilíaco y al músculo psoas (figura 2A). El tumor retroperitoneal izquierdo (figura 2B) medía 4 x 2 cm y era de aspecto y consistencia adiposa, localizado a nivel de L5-S1-S2, adherido al agujero obturatriz.
La paciente evolucionó favorablemente, con remisión parcial del dolor, siendo dada de alta a los tres días.
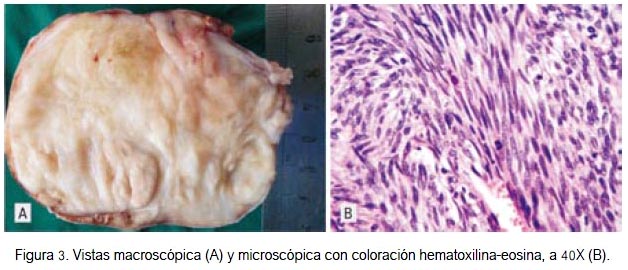
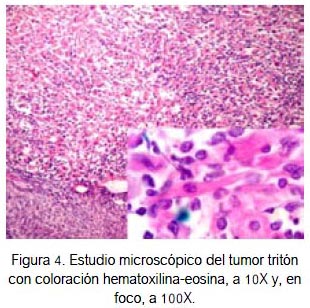
En anatomía patológica, el estudio macroscópico (figura 3A) mostró tumoración sólida con áreas nodulares, a predominio fibrótica. A la microscopia con tinción de hematoxilina-eosina se observó gran celularidad fusiforme con numerosas mitosis (figura 3B), característico de las neoplasias sarcomatosas, como es el tumor maligno de la vaina de los nervios periféricos (7). En otros campos se halló zonas de diferenciación rabdomioblástica (figura 4), incluso con células maduras que mostraban estriaciones transversales.
Por los hallazgos clínicos, quirúrgicos y anátomopatológicos, se concluyó con el diagnóstico de neoplasia maligna de la vaina de los nervios periféricos con diferenciación rabdomiosarcomatosa: tumor maligno de tritón.
DISCUSIÓN
Como se ha señalado en la introducción, los elementos de rabdomiosarcoma dentro de los tumores malignos de la vaina de los nervios periféricos (MPNSTs) fueron descritos por Mas-son en pacientes con neurofibromatosis (NF-1) (1,2). El nombre de tritón, fue usado primero por Woodruff y col (3), y se basó en los miembros supernumerarios inducidos a crecer sobre las espaldas de las salamandras tritón, por trasplante.
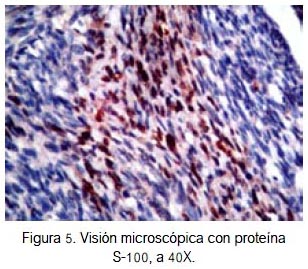
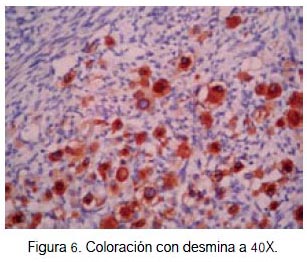
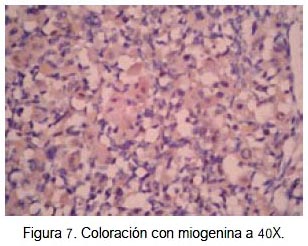
En el caso que presentamos, se identificó rabdomioblastos y células típicas de schwannoma maligno (figura 4). Además, la inmunohistoquímica fue positiva para proteína S-100 (figura 5), desmina (figura 6) y miogenina (figura 7), confirmando el diagnóstico de MTT. Tales criterios concuerdan con los hallazgos histológicos e inmunohistoquimicos de un tumor de tritón.
Epidemiológicamente, la edad media de los pacientes con MTT se ha estimado en 31,7 años (6,8), aunque la enfermedad puede afectar a cualquier persona entre las edades de 0 y 81 años. Parece haber una distribución igual por sexo, y el tumor convive con NF-1 en 44% a 69% de los casos. Cuando se asocia con NF-1, el MTT tiende a presentarse a una edad más temprana. El MTT es un tumor muy agresivo, con tasas bajas de supervivencia (tasa estimada de supervivencia en cinco años de 26%) (6) y tasas altas de metástasis de 48%; la recurrencia local ocurre en 43% (3,9). Los principales factores que afectan la supervivencia parecen ser la ubicación del tumor y la extensión de la exéresis (3,4,6,9,10). Los pacientes con neoplasias de cabeza y cuello o extremidades sobreviven por más tiempo que aquellos con tumores del glúteo, tronco o retroperitoneo (6). La resección completa parece estar asociada con un mejor pronóstico, disminución de las tasas de recurrencia local y metástasis, y una mejor respuesta a terapias adyuvantes; sin embargo, la mayoría de los pacientes con MTTs muere incluso después de recibir todos los tratamientos disponibles, generalmente en el plazo de meses (6,9). El presente caso fue una paciente mujer de 29 años de edad, con antecedente de NF-1. El tumor era enorme, 10 x 14 cm, ubicado en el retroperitoneo pélvico, lado derecho, y el segundo tumor del lado izquierdo más pequeño, que midió 7 x 6 cm, cuyo resultado anatomopatológico fue neurofibroma.
Hasta la fecha, aproximadamente 170 casos de MTT han sido reportados en la literatura mundial (6,11,12). Entre estos, el MTT ubicado en el retroperitoneo es extremadamente raro; utilizando MEDLINE encontramos solo nueve casos publicados en la literatura inglesa; todos los pacientes presentados tuvieron una enorme masa retroperitoneal, que oscilaba entre 7 y 19 cm de diámetro. El tamaño de presentación puede explicarse por el hecho de que los tumores retroperitoneales son a menudo asintomáticos en las primeras etapas, como se muestra en nuestro caso (13). La presencia de un tumor retroperitoneal solo puede expresarse por el inicio insidioso de los síntomas, que son inespecíficos, tales como dolor abdominal vago debido a compresión o infiltración de vísceras adyacentes, vasos o nervios (13,14). Así, los MTTs retroperitoneales se asocian con un peor pronóstico, mayores tasas de enfermedad metastásica y tasas altas de recurrencia, debido a retrasos en el diagnóstico, similar a los MPNSTs en la cavidad abdominal (15,16).
Debido a la rareza del MTT, las modalidades de tratamiento -especialmente en casos avanzados y metastásicos- no están bien establecidas, y no existen pautas estandarizadas de administración. Como en muchos sarcomas de tejidos blandos (STS), la estrategia terapéutica más eficaz para STS retroperitoneal es la resección completa del tumor. El estado de los márgenes microscópicos después de la resección tiene un impacto significativo en los resultados locales y la supervivencia (17). Enneking y col (17) describieron cuatro tipos de márgenes histológicos: intralesional, marginal, amplio y radical. Sin embargo, los márgenes quirúrgicos son difíciles de definir cuando se producen metástasis dentro del abdomen. Además, el espacio retroperitoneal es uno de los sitios más difíciles técnicamente. Las restricciones anatómicas limitan la capacidad para lograr la extirpación amplia en este lugar, que es poco probable que se alcance en la mayoría de los casos. Esto crea el reto de equilibrar la necesidad de márgenes adecuados de control tumoral óptimo con la necesidad de reducir la morbilidad y la pérdida de función, similar a otros STSs (18). El papel de la terapia adyuvante, por ejemplo la radioterapia y la quimioterapia, está muy bien definido y no ha probado ser eficaz (5,9,10), aunque puede ser de valor en los pacientes sometidos a resección completa (19-22). Varios informes sugieren que la resección repetida combinada con quimioterapia o radioterapia puede prolongar la supervivencia (19-21). En nuestra revisión, solo un paciente experimentó la supervivencia a largo plazo (más de 5 años), donde se realizó una resección completa seguida de radioterapia y quimioterapia postoperatoria. Además, la quimioterapia neoadyuvante en TMM ha sido reportada en dos pacientes con enfermedad metastásica (23), aunque el número de casos es demasiado pequeño para permitir una conclusión definitiva sobre la efectividad de la quimioterapia neoadyuvante.
REFERENCIAS BIBLIOGRÁFICAS
1. Ducatman BS, Scheithauer BW. Malignant peripheral nerve sheath tumors with divergent differentiation. Cancer. 1984;54:1049–57. doi:10.1002/1097-0142(19840915)54:6<1049::AIDCNCR2820540620>3.0.Co;2-1. [ Links ]
2. Masson P. Recklinghausens neurofibromatosis sensory neuromas and motor neuromas. Libman Anniversary Volume 2. New York: The International Press; 1932:793–802.
3. Woodruff JM, Chernik NL, Smith MC, Millett WB, Foote FW. Peripheral nerve tumors with rhabdomyosarcomatous differentiation (malignant"Triton" tumors) Cancer. 1973;32:426–39. doi: 10.1002/1097-0142(197308)32:2<426::AIDCNCR2820320221>3.0.Co;2-W. [ Links ]
4. Daimaru Y, Hashimoto H, Enjoji M. Malignant "triton" tumors: a clinicopathologic and immunohistochemical study of nine cases. Hum Pathol. 1984;15:768–78. doi: 10.1016/S0046-8177(84)80169-0. [ Links ]
5. Stasik CJ, Tawfik O. Malignant peripheral nerve sheath tumor with rhabdomyosarcomatous differentiation (malignant triton tumor) Arch Pathol Lab Med. 2006;130:1878–881. [ Links ]
6. Yakulis R, Manack L, Murphy AI. Postradiation malignant triton tumor. A case report and review of the literature. Arch Pathol Lab Med. 1996;120:541–8. [ Links ]
7. Coindre JM. Grading of soft tissue sarcomas: review and update. Arch Pathol Lab Med. 2006;130:1448–53. [ Links ]
8. Haddadin MH, Hawkins AL, Long P, Morsberger LA, Depew D, Epstein JI, Griffin CA. Cytogenetic study of malignant triton tumor: a case report. Cancer Genet Cytogenet. 2003;144:100–5. doi: 10.1016/S0165-4608(02)00935-4. [ Links ]
9. Brooks JS, Freeman M, Enterline HT. Malignant "Triton" tumors. Natural history and immunohistochemistry of nine new cases with literature review. Cancer. 1985;55:2543–9. doi: 10.1002/1097-0142(19850601)55:11<2543::AIDCNCR2820551105>3.0.Co;2-4. [ Links ]
10. Victoria L, McCulloch TM, Callaghan EJ, Bauman NM. Malignant triton tumor of the head and neck: A case report and review of the literature. Head Neck. 1999;21:663–70. doi: 10.1002/(SICI)1097-0347(199910)21:7<663::AID-HED12>3.0.Co;2-4. [ Links ]
11. Terzic A, Bode B, Gratz KW, Stoeckli SJ. Prognostic factors for the malignant triton tumor of thehead and neck. Head Neck. 2009;31:679–88. doi: 10.1002/hed.21051. [ Links ]
12. Woodruff JM, Perino G. Non-germ-cell or teratomatous malignant tumors showing additionalrhabdomyoblastic differentiation, with emphasis on the malignant Triton tumor. Semin Diagn Pathol.1994;11:69–81. [ Links ]
13. Hoshimoto S, Morise Z, Takeura C, Ikeda M, Kagawa T, Tanahashi Y, Okabe Y, Mizoguchi Y, Sugioka A. Malignant Triton tumor in the retroperitoneal space associated with neurofibromatosis type 1: a case study. Rare Tumors. 2009;1:e27. [ Links ]
14. Rekhi B, Jambhekar NA, Puri A, Agrawal M, Chinoy RF. Clinicomorphologic features of a series of 10 cases of malignant triton tumors diagnosed over 10 years at a tertiary cancer hospital in Mumbai, India. Ann Diagn Pathol. 2008;12:90–7. doi: 10.1016/j.anndiagpath.2007.04.010. [ Links ]
15. Radovanovic D, Vukotic-Maletic V, Stojanovic D, Lalosevic D, Likic I, Stojsic Z, Bacetic D. Retroperitoneal "Triton" tumor. Hepatogastroenterology. 2008;55:527–30. [ Links ]
16. DAgostino AN, Soule EH, Miller RH. Primary malignant neoplasms of nerves (malignant neurilemomas) in patients without manifestations of multiple neurofibromatosis (Von Recklinghausens disease) Cancer. 1963;16:1003–14. doi: 10.1002/1097-0142(196308)16:8<1003::AIDCNCR2820160807>3.0.Co;2-S. [ Links ]
17. Enneking WF, Spanier SS, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin orthop Relat Res. 1980;153:106–20. [ Links ]
18. Grimer R, Judson I, Peake D, Seddon B. Guidelines for the management of soft tissue sarcomas. Sarcoma. 2010;2010:506182. [ Links ]
19. Koutsopoulos AV, Mantadakis E, Katzilakis N, Lagoudaki ED, de Bree E, Stiakaki E, Kalmanti M. Long-term survival of a patient with a neurofibromatosis Type 1 associated retroperitoneal malignant triton tumor after multi-modality treatment. ClinNeuropathol. 2011;30:333–5. [ Links ]
20. Kostler WJ, Amann G, Grunt TW, Singer CF, Schneider SM, Brodowicz T, Tomek S, Zielinski CC. Recurrent malignant Triton tumour: first report on a long time survivor. Oncol Rep. 2003;10:533–5. [ Links ]
21. Lang-Lazdunski L, Pons F, Jancovici R. Malignant "Triton" tumor of the posterior mediastinum: prolonged survival after staged resection. Ann Thorac Surg. 2003;75:1645–8. doi: 10.1016/S0003-4975(02)04825-7. [ Links ]
22. Okur FV, Oguz A, Karadeniz C, Citak C, Bayik P, Boyunaga o. Malignant triton tumor of the pelvis in a 2-year-old boy. J Pediatr Hematol oncol. 2006;28:173-6. doi: 10.1097/01. mph.0000201419.05182.29. [ Links ]
23. Thoennissen NH, Schliemann C, Brunnberg u,Schmidt E, Staebler A, Stegger L, Bremer C, Schleicher C, Mesters RM, Muller-Tidow C, Berdel WE. Chemotherapy in metastatic malignant triton tumor:report on two cases. oncol Rep. 2007;18:763–7. [ Links ]
Artículo recibido el 17 de setiembre de 2013 y aceptado para publicación el 9 de enero de 2014.
Los autores señalan no existir conflictos de interés.
Correspondencia:
Correo electrónico: hlimaylla@hotmail.com
