










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkAnales de la Facultad de Medicina
versión impresa ISSN 1025-5583
An. Fac. med. vol.77 no.3 Lima jul./set. 2016
CASOS CLÍNICOS
Neurofibroma cervical pediátrico. Reporte de caso
Pediatric cervical neurofibroma. A case report
Juan Francisco Oré Acevedo1; Martín La Torre Caballero1; Rosmery Urteaga Quiroga1
1 Médico de Cirugía de Cabeza, Cuello y Maxilofacial, Instituto Nacional de Salud del Niño, Lima, Perú.
Resumen
Los neurofibromas son tumores benignos frecuentes en la región de la cabeza y cuello, los cuales son referidos al especialista para su tratamiento respectivo. Se reporta el caso de un varón de 14 años con un tumor cervical de 14 cm en su diámetro mayor, el cual desplazaba las estructuras de la línea media debido al tamaño que presentaba. Después de la resección quirúrgica, el estudio de patología lo catalogó como neurofibroma. Sin déficit motor, salvo la pérdida de la sensibilidad del paladar blando, el reflejo nauseoso y con el síndrome de Horner ya presente, el paciente fue dado de alta. Se concluye que el tratamiento de los neurofibromas es eminentemente quirúrgico, siendo ideal la resección total y parcial según las peculiaridades del neurofibroma. Se puede presentar déficit neurológico, de acuerdo al nervio de donde se origina.
Palabras clave. Neurofibroma, Cervical; Pediatría; Cirugía.
Abstract
Neurofibromas are benign tumors of frequent presentation in the head and neck. We report the case of a 14 years old male who presented a cervical tumor 14 cm in length. Despite the size of the tumor and the fact that it displaced the cervical midline structures, the patient remained asymptomatic. The pathology report following surgery was of a neurofibroma. On discharge there were no motor limitations present, but loss of soft palate sensitivity and nauseous reflex, and the previous Horner syndrome. Surgery is the gold standard treatment and total resection is ideal, but debulking is preferred according to the neurofibroma characteristics. Neurological deficit could be expected.
Keywords. Neurofibroma, Cervical; Pediatrics: Surgery.
INTRODUCCIÓN
Se define a los neurofibromas como tumores benignos originados del tejido neural, presentando tanto elementos derivados de este como del tejido conectivo circundante. El neurofibroma corresponde a un tumor dependiente de la cubierta fibrosa del tejido nervioso, a diferencia del Schwannoma, el cual es proliferación de las células de Schwann. Los tumores neurogénicos tienen diversas presentaciones, son únicos o múltiples, y pueden tener origen de las diversas estructuras nerviosas (1-4).
Se dividen en dos tipos, plexíforme y cutáneo. Los plexiformes suelen ser congénitos, pueden ser localmente invasivos y tienen 4 a 5% de probabilidades de malignización. Los cutáneos se presentan típicamente en la etapa prepuberal y no tienen potencial maligno (2,3).
Los tumores neurogemcos son de consistencia sólida y pueden originarse de cualquier tejido nervioso de la región de la cabeza y del cuello. Su localización es predominantemente a nivel del espacio parafaríngeo, seguido de la región cervical. Ocasionalmente pueden presentar el síndrome de Horner, si el origen es la cadena simpática cervical. Su crecimiento es lento y normalmente asintomático. En los casos de gran tamaño es evidente la deformidad estética con o sin compromiso óseo y pueden llegar a comprimir la vía aérea y/o digestiva, con deformidad de las estructuras vecinas, limitación funcional o dolor (2-12).
La tomografía y la resonancia magnética son estudios de elección. En ambas se puede ver la relación con los vasos profundos del cuello, laringe, tráquea y esófago, así como determinar el tamaño tumoral y la existencia de infiltración o cápsula periférica al tumor. La cirugía es el tratamiento de elección, sin dejar de lado el riesgo de morbilidad debido a los diversos nervios que discurren por la región cervical. Debido a la recurrencia significativa, la edad de la cirugía es controversial.
La quimioterapia y la radioterapia no están indicadas en esta patología. La transformación maligna es sumamente rara (2.9,11).
Se presenta este caso debido a que, a pesar del tamaño considerable del tumor, no mostró síntomas y la probabilidad de secuela neurológica. La resección total quirúrgica fue el tratamiento de elección, teniendo como objetivo la reinserción social del paciente sin limitaciones funcionales.
COMUNICACIÓN DEL CASO
Paciente varón de 14 años que acudió por tumor cervical de crecimiento lento durante 2 años, indoloro, sin alteraciones como disfonía, disfagia o disnea; dicha tumoración tenía consistencia firme, límites bien definidos, móvil y sin compromiso de la piel. Además, el paciente presentó desde su ingreso a la institución un síndrome de Horner, evidente por la ptosis palpebral del lado comprometido (figura 1), el cual se mantendría en el postoperatorio. Se denotaba que el tumor dependía del plexo simpático cervical.
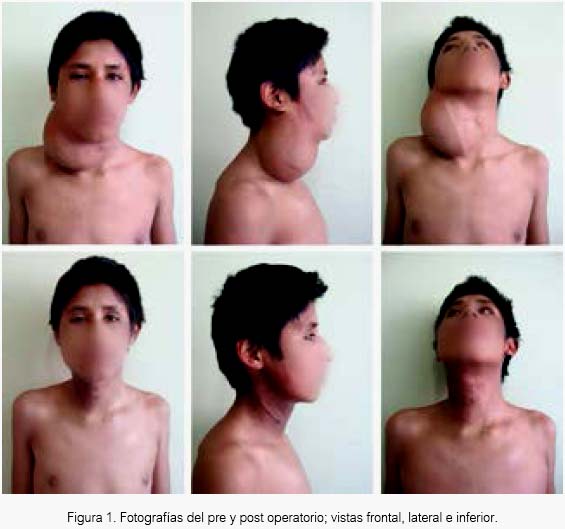
El paciente presentaba cicatriz de una biopsia previa en la institución que lo refirió, biopsia que fue frustra por la profundidad del tumor.
Se le realizó tomografía cervical con contraste para su tratamiento quirúrgico, mostrando una tumoración cervical derecha de aproximadamente 10 cm, la cual no infiltraba las estructuras vecinas, pero desplazaba la vía aérea laringo-traqueal. En las imágenes se apreció una cápsula definida periférica al tumor de contenido heterogéneo, ausencia de calcificaciones, avascular y sin destrucción o infiltración de estructuras vecinas.
Dicha tumoración se localizaba por delante de los cuerpos vertebrales, desde la parte superior hasta el cuello del cóndilo mandibular y la fosa supraclavicular, estando ubicada por dentro de la rama mandibular (figura 2).
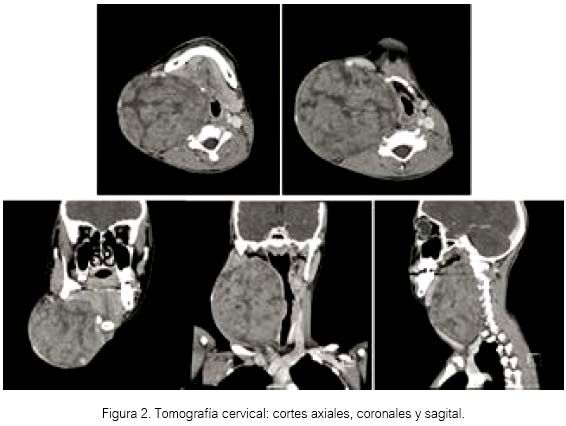
Con el diagnóstico de tumor cervical, probable neurofibroma, por su localización y característica tomo gráfica, se procedió a la cirugía.
Bajo anestesia general se realizó la disección hasta la aponeurosis cervical profunda con disección del paquete vasculonervioso cervical (arteria carótida, vena yugular interna y nervio neumogástrico), logrando la resección del neurofibroma, tumor sólido de consistencia dura, forma ovalada y sin abundante vascularización (figura 3).
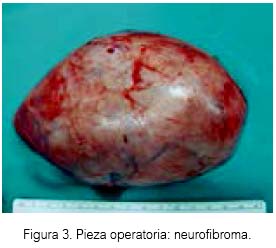
Durante el postoperatorio inmediato, al examen físico no se evidenció lesión de los pares craneales motores (facial, neumogástrico, espinal e hipogloso), nervio frénico simpático cervicalo de los troncos del plexo braquial. Presentó anestesia a nivel del paladar blando ipsilateral, con pérdida del reflejo nauseoso a la palpación, y manteniéndose presente el síndrome de Horner. Debido a la evolución adecuada, fue dado de alta. No ha existido recidiva de la tumoración a los tres años de control postoperatorio.
DISCUSIÓN
Los neurofibromas en cabeza y cuello son tumores benignos dependientes de los pares craneales o de los nervios periféricos en dicha región. Pueden llegar a alcanzar grandes dimensiones, sin producir déficit neurológico, siendo más llamativa la deformidad facial o cervical por la extensión del tumor mismo.
Como los neurofibromas son dependientes de los nervios con función motora, predominantemente, como secuela pueden presentar limitación funcional específica, requiriendo terapia física en el postoperatorio u otro tratamiento quirúrgico secundario.
El tratamiento de los neurofibromas es exclusivamente quirúrgico, tratando de establecer antes de la cirugía, de ser posible, el nervio del cual se origina o es dependiente. La resección completa es el tratamiento ideal. Sin embargo, en ocasiones no es posible debido al déficit funcional significativo que ocasionaría, en cuyo caso se podría aceptar resecciones parciales.
REFERENCIAS BIBLIOGRÁFICAS
1. Elden L, Zur K. Congenital malformations of the head and neck. Philadelphia: Springer; 2014. [ Links ]
2. Rahbar R, Rodríguez-Galindo C, Meara J. Pediatric head and neck tumors. New York: Springer; 2014. [ Links ]
3. Bagheri SC, Bell RB, Khan HA. Current therapy in oral and maxillolacial surgery. Missouri: Elsevier; 2012. [ Links ]
4. Cohen JI, Clayman GL. Atlas of head & neck surgery. 1st Ed. Philadelphia: Elsevier; 2011. [ Links ]
5. De Souza C. Atlas of head & neck surgery. 1 st Ed. India: Jaypee; 2013. [ Links ]
6. Pérez J, Martin F. Tumores neurogénicos benignos del cuello. Rev Chilena Cirugía. Abril 2001 ;53(2):214-9. [ Links ]
7. Navarro C. Tratado de cirugía oral y maxilolacial. 1ra Ed. Madrid: Arán; 2004. [ Links ]
8. Cantini JE, Prada JR. Cirugía craneolacial. 1ra Ed. Bogotá: Impresión Médica. 2012. [ Links ]
9. Shah J, Patel S. Cirugía y oncología de cabeza y cuello. 3ra Ed. Madrid: Elsevier; 2004. [ Links ]
10. Eisele DW, Smith RV. Complications in head and neck surgery. 2nd Ed. Philadelphia: Elsevier; 2009. [ Links ]
11. Harrison LB, Sessions RB Hong WK. Head and neck cancer. 2nd Ed. Philadelphia: Lippincott Williams & Wilkins; 2004. [ Links ]
12. Myers EN, Suen JS. Cancer of the head and neck. 4th Ed. Philadelphia: Saunders; 2003. [ Links ]
Conflicto de interés: Los autores declaran no tener conflicto de interés.
Correspondencia:
Dr. Juan Francisco Oré Acevedo
Dirección: Calle Francisco de Orellana Mz R, Lote 23, Opto 301, Sta. Patricia, La Molina, Lima
Teléfono: 993 464 995
Correo electrónico: juanfcoore@yahoo.com
Articulo recibido el 17 de marzo de 2016 y
Aceptado para publicación el 27 de junio de 2016.
