











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
El término de Queratoquiste Odontogénico (QQO) se utilizó por primera vez en la década de 1950 para describir todos los quistes odontogénicos que contienen queratina, inicialmente algunos patólogos denominaban erróneamente a esta entidad como quiste primordial; la denominación de QQO fue adoptada en la clasificación de la Organización Mundial de la Salud de 1992. En el 2005 se reclasificó esta lesión como neoplasia cambiando el nombre a Tumor Odontogenico Queratoquistico (TOQ) debido a la alta tasa de recurrencia, comportamiento clínico agresivo, asociación con el Síndrome de Carcinoma Células Basa-les Nevoides (SCCBN) o Síndrome de Gorlin-Goltz (SGG) y mutaciones en el gen supresor de tumores Patched (PTCH)1. Además, el año 2007 Daley et. al. publicaron un caso de un TOQ sólido, lo que viene a reforzar el concepto de la naturaleza neoplásica de dicha patología 2.
La clasificación de 2017 volvió a la terminología original y bien aceptada de QQO porque muchos artículos mostraron que la mutación del gen PTCH podría encontrarse en lesiones no neoplásicas, incluyendo quistes dentígeros, además, muchos investigadores sugirieron que la resolución del quiste después de la marsupialización no fue compatible con un proceso neoplásico1.
La presencia QQOs múltiples generalmente está relacionado con el SGG3,4, aunque también se ha encontrado asociado a otros síndromes, como el de Simpson-Golabi-Behm5, síndrome oro-facio-digital6, síndrome de Noonan7, o el síndrome de Ehler-Danlos8. Sin embargo, se ha encontrado en la literatura reportes en los que se menciona la presencia de QQOs múltiples en pacientes no sindrómicos9,10,11. El término “múltiple” se refiere a los antecedentes de vida del paciente y no a la aparición de varios quistes presentes en un momento dado. Los QQOs múltiples significan la aparición de estos quistes a lo largo de la vida, y solo el 5% de estos casos se han notificado en individuos no sindrómicos11,12. El SGG es un desorden autosómico dominante, el primer caso fue descrito por Robert James Gorlin y William Goltz en 1960 con anomalías esqueléticas, presencia de múltiples quistes en maxilares y carcinoma de células basales. Este síndrome posee una penetrancia completa y expresividad variable, debida a mutaciones germinales en los genes PTCH1 (locus 9q22.32) o SUFU (locus 10q24.32) involucrados en la vía molecular Sonic Hedhehog (SHH) relacionada con funciones como la embriogénesis, carcinogénesis y reparación de tejidos. La alteración de esta vía explicaría las características clínicas del SGG, siendo las anomalías congénitas consecuencia de la mutación germinal del gen PTCH1, mientras que el desarrollo de los carcinomas basocelulares y otros tu-mores se deben a la segunda mutación que se produce en el otro alelo. Los pacientes que porten estas mutaciones, tienen una probabilidad del 50% de heredarla a su descendencia. El 70-80% de los casos de SGG, se debe a que el paciente heredó la mutación de alguno de sus padres, mientras que el 20-30% restante corresponden a casos “de novo” o aislados en la familia13,14. La prevalencia de este síndrome varía de 1: 30,000 a 1: 256,000 según las conclusiones de diferentes estudios14, y tiene una incidencia tanto esporádica como familiar, afecta a ambos sexos y se observa durante la primera, segunda y tercera décadas de la vida15,16.
Existe una prevalencia ligeramente más alta de QQOs esporádicos en hombres que en mujeres, con una pro-porción de 1:1.42. Pero en el caso de QQOs múltiples asociados con el síndrome es de 1:112. Los QQOs pueden presentarse como el primer signo de SGG, y pueden identificarse en pacientes menores de 10 años17.
El propósito de este reporte de caso es proporcionar una base objetiva para el manejo terapéutico de los QQOs en pacientes con SGG y una revisión de la literatura científica.
Reporte de caso
Paciente de sexo femenino de 63 años, mestiza y procedente de Lima, con antecedente de SGG, sometida a múltiples intervenciones quirúrgicas desde los 23 años de edad: exceresis de múltiples nevos basocelulares en región frontal, labial y orbitaria, realizadas por los servicios de Cirugía de Cabeza y Cuello, Dermatología, y Oftalmología; y exodoncias, enucleación y curetaje de QQOs múltiples en los cuatro cuadrantes de ambos maxilares (todas las lesiones fueron confirmadas mediante estudio anatomopatologico), realizados el año 2007 por el servicio de Cirugía Bucomaxilofacial. A los dos años se reporta recidiva de los QQOs en el maxilar superior izquierdo y cuerpo con extención a rama mandibular derecha, por lo que es sometida nuevamente a enucleación y curetaje, además de ostectomia periférica de estas lesiones.
El servicio de Genética Médica, elabora el heredograma (figura 1), indicando que no cuenta con antecedentes familiares, además, presenta defectos congénitos óseos, y concluye que el cuadro clínico es compatible con SGG, que es originado por gen autosómico dominante, probable mutación de novo (no hay hermanos afectados); asimismo, concluye que para detectar el gen se requiere estudios de biología molecular que no se realizan en el país.
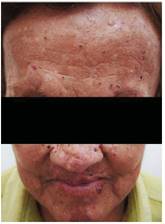
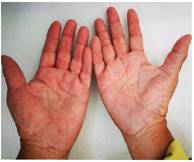
Al examen físico se evidencia fascie característica, prominencia frontal, parietal y arcos superciliares, hipertelorismo, puente nasal ancho, cicatrices cutáneas múltiples en regiones frontal, orbitaria, nasal, labial, y mentoniana, secuela de exéresis de nevos basocelulares, hiperpigmentaciones faciales múltiples, de bordes irregulares (figura 2); en palmas de ambas manos se identificaron múltiples pits (figura 3).
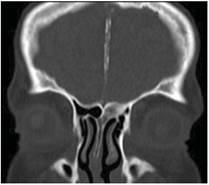
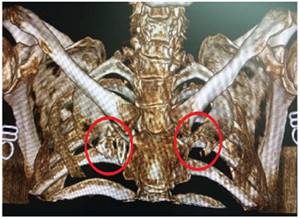
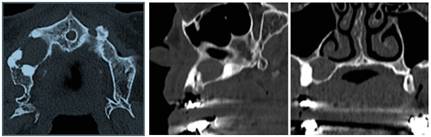
La tomografía helicoidal multicorte revela calcificación de la hoz del cerebro (figura 4), costilla bífida (figura 5). A nivel de los maxilares se observa imagen isodensa localizada en maxilar superior derecho, de forma ovalada, bordes definidos y corticalizados, presentando el adelgazamiento de la cortical ósea vestibular. Descripción imagenológica compatible con QQO (figura 6).
Teniendo en cuenta los antecedentes patológicos de la paciente, se programa para intervención quirúrgica en sala de operaciones, con el diagnostico presuntivo de QQO recurrente (9 años después de la última intervención quirúrgica); se realiza la enucleación y curetaje de la lesión, además de ostectomia periférica del hueso adyacente, finalmente se aplica solución de Carnoy durante 3 minutos en el lecho quirúrgico, para prevenir nueva recurrencia (Figura 7).
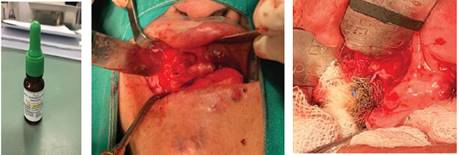
Se envía la muestra al Departamento de Anatomía Patológica del Hospital Nacional Edgardo Rebagliati Martins para el análisis correspondiente, el estudio histológico confirmó que el diagnóstico definitivo fue Queratoquiste Odontogénico (Figura 8).

La paciente se encuentra en el sexto mes post operatorio, presenta ausencia de sintomatología, y a la evaluación imagenológica se evidencia disminución de tamaño del lecho quirurgico en un 40 %.
DISCUSIÓN
La asociación del QQO con el SGG permitió iniciar investigaciones genéticas en el QQO, el SGG es una enfermedad que predispone al cáncer y en el año 1996 se descubrió que en su etiología interviene la pérdida funcional del gen PTCH16. El gen PTCH1 es un gen supresor de tumores, lo que significa que evita que las células proliferen demasiado rápido o de manera descontrolada; las mutaciones en este gen impiden la producción de PTCH1 o conducen a la producción de una versión anormal del receptor. Un receptor PTCH1 alterado o faltante no puede suprimir eficazmente el crecimiento y la división celular; como resultado, las células proliferan incontrolablemente para formar los tumores que son característicos del SGG18.
El QQO puede tener un comportamiento similar al de una neoplasia maligna ya que puede penetrar en la órbita y la fosa infratemporal. La tasa de crecimiento puede variar entre 2 a 14 mm por año, lo que se considera como un crecimiento rápido para este tipo de lesiones. Este comportamiento es más frecuente cuando esta lesión se asocia al SGG. Si se compara el índice mitótico de los QQOs con el resto de quistes los resultados son que en éstos el porcentaje de mitosis es elevado (40%), mientras que en quistes dentígeros el porcentaje de proliferación sería un 17% y en los quistes radiculares un 15,5 % 19.
Variantes heterocigotas patógenas en los genes PTCH1, PTCH2 y SUFU están asociados con el SGG, y su fenotipo puede variar con más de 100 anomalías asociadas a este síndrome18. Hasta la fecha, se han notificado más de 230 mutaciones en la línea germinal de PTCH1 asociadas con SGG. Las mutaciones del gen PTCH se han documentado en hasta al 85% de los QQO sindrómicos y alrededor del 30% de los QQO no sindrómicos20.
La patogenia del SGG aún no se ha aclarado. La correlación genotipo-fenotipo no se ha establecido, y no se ha determinado ninguna correlación entre el tipo y la posición de la mutación dentro del gen PTCH1 con ciertas características de la enfermedad, como el número y la edad de inicio de carcinoma de células basales. Además, los pacientes con mutaciones idénticas difirieron en la extensión de los hallazgos clínicos 21.
Los QQOs muestran un mecanismo diferente de crecimiento y comportamiento biológico. Aunque se cree ampliamente que los quistes odontogénicos se expanden como consecuencia del aumento de la presión osmótica dentro del lumen del quiste, este mecanismo no parece ser correcto para el QQO, su crecimiento puede ser asociado a factores desconocidos en el epitelio en sí o actividad enzimática en la pared fibrosa, por eso algunos investigadores sugieren que debe ser considerado como una neoplasia quística benigna en lugar de un quiste 22.
Los métodos de inmunohistoquímica utilizando marcadores de proliferación celular demuestran que la proliferación celular desempeña un importante papel en el desarrollo del QQO. En las células epiteliales de los QQOs se ha observado una inmunolocalización aumentada de la proteína p53, del receptor del factor de crecimiento epidérmico (EGFr), del antígeno nuclear de la proliferación celular (PCNA) y del antígeno Ki-67 16.
Se han realizado estudios comparativos de los QQOs asociados y no asociados al SGG, y se encontró la presencia de mayor número de quistes satélites, proliferaciones sólidas del epitelio, inflamaciones, calcificaciones y más intensa actividad mitótica de las células epiteliales de los QQOs asociados al SGG16, además, los QQOs sindromicos se dan en individuos más jóvenes, son múltiples, comúnmente en la región maxilar posterior, epitelio más delgado y mayor recurrencia (82%), a diferencia que los QQO no sindromicos (61%)11,23.
La recurrencia del QQO puede desarrollarse de 3 formas diferentes: mediante la eliminación incompleta del revestimiento del quiste original; por retención de quistes satélites, de microquistes o islas epiteliales en la pared del quiste original; o como nuevos QQOs de los brotes epiteliales de la capa basal del epitelio oral11,12.
Se han descrito más de cien aspectos clínicos del SGG, pero no siempre se encuentran en todos los pacientes16. El estándar de atención actual establece el diagnóstico del SGG mediante el cumplimiento de los criterios clínicos diagnósticos existentes, mientras que la prueba de diagnóstico de oro es genética. El SGG debe ser altamente sospechoso si el paciente tiene dos criterios mayores y un criterio menor o uno mayor y tres criterios diagnósticos menores, de acuerdo a los parámetros propuestos por Evans et al., y modificados por Kimonis et al., en 197324. Nuestra paciente presentaba cinco criterios mayores y un criterio menor (Tabla 1), la edad a la que fue diagnosticada de SGG fue aproximadamente a los 23 años de edad, que coincide con la media para pacientes “de novo”, grupo al cual pertenece14, y el QQO recurrente se encontraba ubicado en el maxilar superior (más común en QQOs asociados a SGG).
Tabla 1 Criterios para diagnosticar síndrome de Gorlin-Goltz
| Criterios | Paciente |
|---|---|
| Criterios Mayores | |
| Más de 2 CCB o un CCB en pacientes menores de 20 años de edad | Presentes |
| QQO de la mandíbula (comprobados mediante análisis histológico) | Presentes |
| Tres o más pits palmar o plantar | Presentes (palmar) |
| Calcificación bilamelar de la hoz del cerebro | Presente |
| Costillas bífidas, fusionadas o marcadamente extendidas | Presentes (costillas bífidas) |
| Un familiar de primer grado con CNCB | Ningún familiar |
| Criterios Menores | |
| Macrocefalia | No determinado |
| Malformaciones congénitas (p. Ej., Labio o paladar hendido, protu-berancia frontal, cara gruesa o hipertelorismo moderado o grave) | Presente (prominencia frontal, hipertelorismo, cara gruesa) |
| Otras anomalías esqueléticas (p. Ej., Deformidad de sprengel, marcada deformidad del pecho y marcada sindactilia) | No determinado |
| Anomalías radiológicas (p. Ej., Puente de la silla turca, anomalías ver-tebrales, defectos oseos en manos y los pies) | No determinado |
| Fibroma ovárico o meduloblastoma | No determinado |
| CCB: Carcinoma de celulas basales; CNCB: Carcinoma nevoide de celulas basales; QQO: Queratoquistes odontogenicos |
Si las características clínicas no son concluyentes, se usan las pruebas genéticas para identificar una mutación de línea germinal heterocigótica en PTCH1, si el paciente tiene un resultado negativo para PTCH1, se deben considerar las pruebas de SUFU y PTCH214. No existe una prueba de laboratorio específica para diagnosticar el SGG, los pacientes afectados pueden tener niveles altos de monofosfato de adenosina cíclico y diuresis con fosfato alterada en la prueba de parathormona25.
A lo largo de los años, se han propuesto diferentes técnicas quirúrgicas para los QQO. A grandes rasgos, las modalidades de tratamiento se dividen en conservadoras y radicales. Los métodos conservadores incluyen: enucleación simple con o sin curetaje o marsupialización /descompresión, con o sin medidas terapéuticas secundarias. Los métodos agresivos incluyen la ostectomía periférica, curetaje químico con solución de Carnoy, crioterapia, electrocauterio, y resección (en bloque o marginal)11,26.
La tasa de recurrencia de QQOs esporádicos fue del 17,6% después de la enucleación y el 22,2% después del tratamiento combinado de marsupialización y enucleación, mientras que para los QQOs sindrómicos las tasas de recurrencia fueron 25% y 68.8% respectivamente. En cuanto al efecto del tratamiento óseo, el 71,4% de los casos esporádicos de QQOs tratados con ostectomia periferica no tuvieron recurrencia, pero para los QQOs síndromicos el tratamiento óseo fue menos efectivo, ya que solo el 47.6% no tuvo recurrencia22.
En el caso presentado el tratamiento realizado fue la enucleación, seguida de ostectomia periférica y aplicación de solución de Carnoy. En el estudio de Kaczmarzyk et. al. reportan 0% de recurrencia a los 5.25 años de seguimiento, en este tipo de tratamiento27. Pogrel realiza la ostectomia periférica previa tinción de azul de metileno los márgenes óseos, todo el hueso residual teñido de azul se elimina con una fresa.
Dado que el azul de metileno mancha el hueso a una profundidad de aproximadamente 0,5 mm en hueso cortical y 1-1,5 mm en hueso esponjoso, cualquier residuo de las células dentro de esta área pueden ser detectadas y eliminadas28. El uso de la solución de Carnoy (alcohol absoluto: fijador que endurece el tejido por contracción; cloroformo: aumenta la velocidad de fijación; ácido acético glacial: hace que el tejido se hinche y evite el endurecimiento; y cloruro férrico), subsecuente a la enucleación quística, per-mite destruir los quistes satélites alojados en el mar-gen óseo, y así prevenir las recurrencias. Tiene una profundidad de penetración ósea media de 1.54 mm,11,12; y existen dudas con respecto a las posibles complicaciones resultantes de su uso, tales como infección, dehiscencia, formación de secreciones óseas y neuropatía22.
Actualmente se está promoviendo el uso de agentes inhibidores de la vía de señalización de Hedgehog (HH), aprobados por la FDA, como el Vismodegib, con buenos resultados para el tratamiento del carcinoma basocelular y QQO en pacientes con SGG. Además, se ha encontrado que los antiinflamatorios no esteroideos (Celecoxib) tienen un efecto quimio-preventivo en humanos y ratones con PTCH1 +/− predispuestos genéticamente14.
Finalmente, se recomienda que los QQO esporádicos se deben seguir de cerca durante los primeros 7 años postoperatorios, mientras que los casos síndromicos deben ser controlados durante los primeros 5 años postoperatorios, después de este tiempo los pacientes pueden ser seguidos cada dos años22.

