










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú vol.37 no.4 Lima oct./dic. 2017
ARTÍCULO DE REVISIÓN
Colangitis biliar primaria. Parte 1. Actualización: generalidades, epidemiología, factores involucrados, fisiopatología y manifestaciones clínicas
Primary biliary cholangitis. Part 1. State of the art, epidemiology, physiopathology and clinical manifestations
Diego Andrés Rodríguez Lugo1a, Jorge Julián Coronado Tovar1a, Giovana Alejandra Solano Villamarin1,b, Wiliam Otero Regino1c,2
1 Universidad Nacional de Colombia. Bogotá, Colombia.
2 Hospital Universitario Nacional de Colombia. Bogotá, Colombia.
a Estudiante, b Residente de Medicina Interna, c docente
RESUMEN
La colangitis biliar primaria (CBP), es una colangiopatía crónica caracterizada por la destrucción selectiva de las células epiteliales biliares de conductos hepáticos de pequeño y mediano calibre, que afecta principalmente a mujeres. Los principales síntomas son la fatiga y el prurito, sin embargo, gran porcentaje de los pacientes pueden ser asintomáticos. El diagnóstico se basa en anticuerpos antimitocondriales (AMA) con títulos >1:40, fosfatasa alcalina >1,5 veces del límite superior normal por más de 24 semanas e histología hepática compatible con la patología. Se asocia con múltiples enfermedades principalmente de carácter autoinmune extra hepáticas, enfermedades tiroideas, óseas, entre otras. El tratamiento de primera línea es el ácido ursodesoxicólico (AUDC) que a pesar que no cura la enfermedad, mejora las pruebas del perfil hepático, así como el retraso en la progresión a cirrosis. Actualmente se encuentran en estudio nuevos tratamientos y terapias adyuvantes. El propósito de esta revisión es ofrecer una actualización de este tema que se presenta en los servicios de medicina interna y gastroenterología; para su realización se conformó un equipo interdisciplinario que desarrolló una búsqueda en la base Medline a través de PubMed con las palabras claves correspondientes y se procedió a una lectura crítica y analítica de títulos, resúmenes y textos completos para el filtro, extracción y síntesis de la información encontrada.
Palabras clave: Colangitis; Cirrosis; Prurito; Ictericia (fuente: DeCS BIREME).
ABSTRACT
Primary biliary cholangitis (PBC) is a chronic autoimmune cholangiopathy characterized by a selective destruction of biliary epithelial cells of small and medium caliber hepatic ducts, which mainly affects women. The main symptoms are fatigue and pruritus, however, a large proportion of patients may be asymptomatic. The diagnosis is based on AMA titers >1:40, alkaline phosphatase >1.5 times the upper limit for more than 24 weeks and compatible liver histology. It is associated with multiple autoimmune diseases mainly extrahepatic, thyroid diseases, bone diseases, among others. The first line treatment is ursodeoxycholic acid (UDCA), that improves liver function tests and delay the progression to cirrhosis. Currently, there are new treatments and adjuvant therapies on study. The purpose of this review is to offer an update in this topic, which is very important in gastroenterology and internal medicine. We formed an interdisciplinary team to search in the database Medline thorough PubMed with the key words describe below, we made a critical lecture of the titles and abstracts of each article to write this paper.
Keywords: Cholangitis; Cirrhosis; Pruritus; Jaundice (source: MeSH NLM).
INTRODUCCIÓN
La colangitis biliar primaria (CBP), inicialmente llamada cirrosis biliar primaria (1), es una enfermedad hepática crónica, caracterizada por inflamación y destrucción progresiva de los ductos biliares interlobulares, originando colestasis, fibrosis, cirrosis y enfermedad hepática terminal en pacientes no tratados (1,2). Por la participación de mecanismos inmunológicos en la destrucción tisular, clásicamente se le ha considerado una enfermedad autoinmune (1-3). Sin embargo, no tiene las características de una verdadera enfermedad autoinmune, las cuales deben cumplir los criterios de Witebsky (4), que son los siguientes: demostración de linfocitos auto reactivos contra antígenos autólogos, demostración de los correspondientes auto antígenos y finalmente la demostración experimental de que el mencionado proceso autoinmune, produce una enfermedad similar a la de los humanos. En adición, la enfermedad no responde a medicamentos inmunosupresores (1,2,3,5-9).
La presente revisión de la literatura pretende ofrecer una actualización de este tema puesto que es un gran reto para médicos internistas y gastroenterólogos abordar de forma integral esta patología. Para ello, se ofrece una síntesis de la literatura dividida en dos partes: la primera sobre generalidades, epidemiologia, factores de riesgo, y la segunda sobre manifestaciones clínicas, diagnóstico, enfermedades asociadas, tratamiento y pronóstico.
MATERIAL Y MÉTODOS
Se conformó un equipo interdisciplinario que desarrolló una búsqueda de la literatura en la base de datos Medline a través de PubMed. Se definieron los términos de búsqueda así como palabras claves y se realizó un algoritmo de búsqueda por cada sección a realizar: ("Liver Cirrhosis, Biliary"[Mesh] AND "Epidemiology"[Mesh]), ("Liver Cirrhosis, Biliary"[Mesh] AND "Risk Factors"[Mesh]),("Liver Cirrhosis, Biliary"[Mesh] AND "physiopathology" [Subheading]).
La búsqueda se limitó a artículos en inglés. Se incluyeron únicamente ensayos clínicos, ensayos clínicos controlados, meta-análisis, estudios multicéntricos y revisiones. Se usaron los filtros "Humans" y "10 years" para las secciones de factores de riesgo y fisiopatología.
Se excluyeron aquellos escritos en un idioma diferente al inglés y artículos que aportaban información incompleta, así como, estudios que no poseían relación con el objeto de esta revisión.
Teniendo en cuenta los criterios de inclusión y excusión, a cada investigador de forma independiente se le asignaron 3 de las secciones propuestas para la realización de la revisión. Se ejecutó un primer filtro con base en el título y posteriormente un segundo filtro según el resumen. El autor con más experiencia en el tema (WO), determinó finalmente los artículos para una lectura integral y analítica.
RESULTADOS
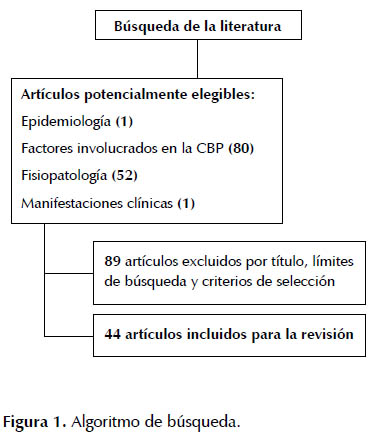
La búsqueda en la literatura para la realización de esta actualización se muestra en la Figura 1. Se presenta la información en 5 secciones: generalidades, epidemiologia, factores involucrados en la CBP, fisiopatología y manifestaciones clínicas.
Generalidades
La CBP fue descrita por primera vez en Londres por Addison y Gull en el año 1851 en pacientes que tenían alteraciones dermatológicas asociadas a enfermedad hepática (10). El nombre original fue "colangiopatía crónica destructiva no supurativa" (1-3), pero por ser muy largo no tuvo aceptación y fue reemplazado por "cirrosis biliar primaria" (1). Este último término fue acuñado por Dauphinee et al. en 1949 y la primera serie de casos fue reportada por Ahrens et al. en 1950 (11). Después de varias décadas, ese último nombre se reemplazó por colangitis biliar primaria, conservando las mismas siglas PBC (Primary Biliar Cholangitis) ó CBP en español (1). La modificación del nombre tuvo lugar en mayo de 2014, en la segunda conferencia monotemática sobre cirrosis biliar primaria, de la Asociación Europea para el estudio de las enfermedades del hígado (EASL) (1). El nombre se cambió porque "cirrosis biliar primaria" es inexacto, ya que la cirrosis, aparece en el estado IV de la enfermedad y no está presente en los estadios iniciales de la misma (1-3,5). Además, no todos los pacientes progresan a cirrosis y menos aun cuando son tratados con ácido ursodeoxicólico (UDCA) (1). Por tanto, los pacientes y sus familiares sentían que eran estigmatizados con una enfermedad (cirrosis) que no tienen, generándoles incertidumbre y angustia. En el nombre actual la palabra cirrosis fue reemplazada por colangitis (2). La nueva denominación ha tenido amplia y rápida aceptación, pero también ha generado críticas (1,12). Se considera que colangitis, denota infección de la vía biliar y además decir colangitis biliar es una "tautología", ya que hace referencia al mismo sitio o tejido y por lo tanto sería una repetición, similar a decir "persona humana". No obstante, esta objeción semántica, el término ya está universalmente incorporado.
Epidemiología
La CBP es una entidad relativamente rara. Tiene una prevalencia de aproximadamente 20 a 40 casos por cada 100 000, afecta más frecuentemente a las mujeres con una relación de 10:1 con respecto a los hombres (7). Aparece entre la quinta y sexta década de la vida (8). La más alta prevalencia se encuentra en Estados Unidos (USA) y en Europa, con una estimación respectiva de 40 y de 20 a 25 casos por cada 100 000 habitantes (10). Aunque esa mayor prevalencia puede reflejar mayor detección en esos países, así como más estudios epidemiológicos, puede ser en realidad un sesgo de detección (11). La incidencia en USA, Europa, Asia y Australia es de 0,9 a 5,8 /100 000 personas/año (10). En Colombia no hay estudios epidemiológicos, pero sí reportes de CBP superpuesta a hepatitis autoinmune (13). Por la baja prevalencia, ha sido considerada una enfermedad huérfana (14). Una enfermedad huérfana se considera la que ocurre con una frecuencia de menos de un caso por cada 2000 personas (15).
Factores involucrados en la CBP
La CBP, es una entidad multifactorial. Para su aparición, se propone la participación de factores genéticos, epigenéticos, medioambientales, infecciosos y más recientemente la microbiota intestinal (1-3,5,6,15- 18). La mayor incidencia ocurre en la peri-menopausia y más frecuentemente cuando hay antecedente de colestasis gestacional (8).
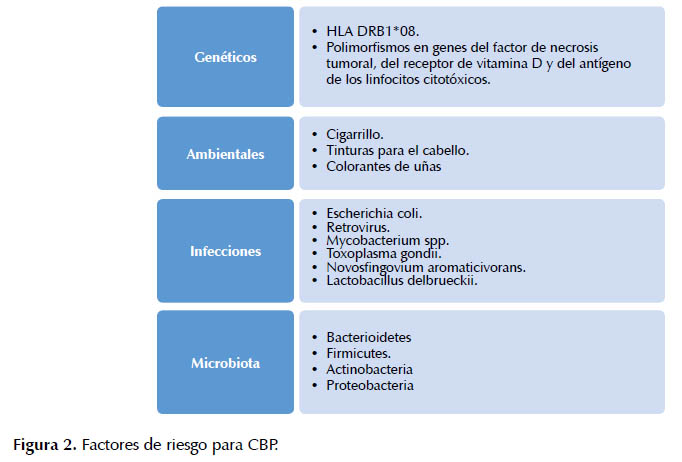
Factores genéticos. Del 1 al 6% de los pacientes con CBP, tienen un familiar de primer grado de consanguinidad afectado por la entidad (8,9). Las hijas de pacientes tienen un riesgo 11 veces mayor de padecer la enfermedad (2). La concordancia entre gemelos monocigóticos es del 63% (8), lo cual destaca la importancia de factores adicionales no genéticos. Diversos polimorfismos tales como DRB1*08, DR3, DPB1*0301 y el DRB1*08-DQA1*0401 – DQB1*04 aumentan el riesgo (2,8,10). Los alelos DRB1*11 y DRB1*13 son factores protectores (2).
Factores ambientales. El cigarrillo es el factor de riesgo más estudiado (8,9). Otros factores de riesgo son las tinturas para el cabello y los esmaltes de uñas (19). Las sustancias tóxicas implicadas producen disminución de la tolerancia inmunológica y favorecen un mayor riesgo de padecer la enfermedad (19-21).
Infecciones. Las infecciones urinarias recurrentes, especialmente por Escherichia coli, se han asociado con un mayor riesgo para la enfermedad (11). Se cree que el mecanismo implicado es la similitud de los antígenos bacterianos con proteínas mitocondriales del individuo, que favorecería reacciones inmunológicas cruzadas entre esas estructuras (8).
Microbiota. La microbiota representa un nuevo "órgano" (22,23). A nivel gastrointestinal (GI), se han descrito más de 50 phyla bacterianas, pero sólo cuatro son las más importantes: Bacteroidetes, Firmicutes, Actinobacteria y Proteobacteria (22). Más de 3000 especies bacterianas residen en el tracto GI y más del 90% de las mismas, pertenecen a Bacteroidetes y Firmicutes (22). Desde el estómago hasta el colon, de manera progresiva, aumenta la densidad bacteriana por cada gramo de contenido intraluminal. En el estómago hay 101<-2, duodeno 103, yeyuno 104, ileon 107 y colon 1022-24. La microbiota tiene múltiples funciones; entre esas la más importante es proteger contra la colonización por patógenos endógenos y exógenos, por diversos mecanismos, como la inhibición de la adhesión de micro-organismos, competir por los nutrientes intra-luminales y modular el sistema inmune y la inflamación, activando macrófagos, neutrófilos, linfocitos y, asimismo, mediar la tolerancia inmunológica y aumentando la producción de diversas citoquinas como la IL-1β e IL-22 (22-24).
Los ácidos biliares (AB) tienen diversas funciones tales como eliminar productos de desecho y catabolitos como el colesterol y las bilirrubinas por las heces, así como, la absorción y digestión de lípidos y vitaminas liposolubles. En adición, son potentes antimicrobianos que inhiben el crecimiento de microorganismos y su adhesión, protegiendo contra infecciones ascendentes hacía la vía biliar. Sin embargo, diversos gérmenes son resistentes a los efectos antimicrobianos de la bilis y modifican la composición de la microbiota intestinal y biliar (25).
La colestasis presente en el CBP podría afectar la microbiota, la susceptibilidad a infecciones y la integridad del epitelio biliar (25). Normalmente, el 95% de los ácidos biliares son reabsorbidos por el epitelio intestinal y trasportados nuevamente al hígado por la vena porta, donde serán re-conjugados y re-excretados con la bilis al duodeno (23). El 5% de los ácidos biliares no reabsorbidos, pueden ser modificados por la microbiota gastrointestinal aerobia y anaerobia. Así, por ejemplo, los ácidos biliares primarios (cólico y quenodeoxicólico) pueden ser transformados a más de 20 diferentes AB secundarios y terciarios por la microbiota anaerobia. La bacteria Ruminococcus gnavus puede formar UDCA, un AB terciario, utilizado para el tratamiento de la CBP (24). Las hidrolasas bacterianas desconjugan los AB primarios taurocolato a colato y este puede alterar diversas funciones sistémicas y hepáticas.
La disminución o ausencia de AB en el tracto intestinal, puede favorecer la aparición de sobrecrecimiento bacteriano y la modificación de la microbiota del colon (25). El hallazgo de respuesta IgM policlonal en CBP, similar a la encontrada en infecciones bacterianas crónicas y la predisposición de los pacientes con CBP a infecciones urinarias recurrentes son argumentos en favor de la relación entre microbios y CBP (16). Los diferentes factores de riesgo, se muestran en la Figura 2.
Fisiopatología
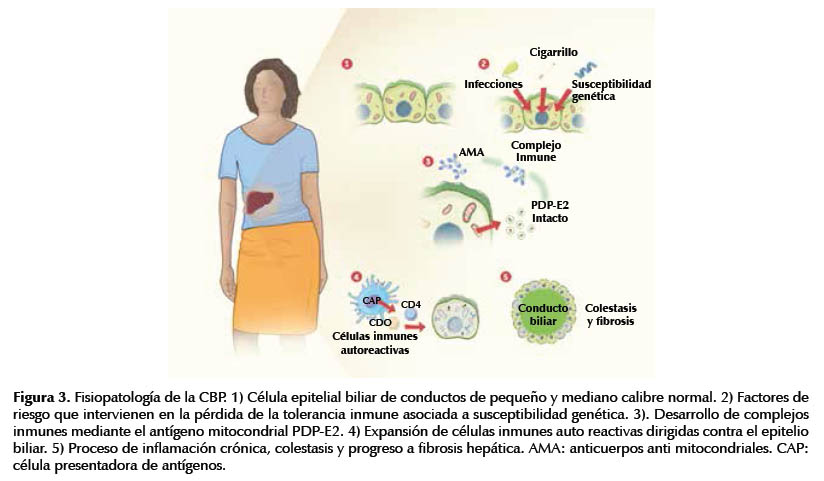
La CBP pertenece al síndrome de ductopenia o conductillos biliares evanescentes (3). Hasta el momento no se conocen con exactitud los mecanismos patogénicos implicados (8). La lesión básica final es la destrucción lenta y progresiva de las células epiteliales biliares (CEB) de los conductos biliares de pequeño y mediano tamaño septales e intralobulillares (5-7). El daño sería consecuencia del ataque inmune a esas estructuras por la pérdida de la tolerancia inmune, alteración de la inmuno-regulación y apoptosis de las CEB por lo cual, histológicamente hay infiltrados linfociticos densos periportales (6,7). La destrucción ductal biliar produce colestasis, que ocasiona la sintomatología de la enfermedad como fatiga, debilitamiento, prurito y conforme avanza la enfermedad, hay aparición progresiva de fibrosis, ictericia y finalmente cirrosis (8- 10). Esta última se encuentra en el 50% de los pacientes cinco años después del diagnóstico (8,9).
Las células efectoras son linfocitos T auto-reactivos ante los dominios lipoil interno y externo de las subunidades E2 del complejo enzimático piruvato – deshidrogenasa (PDC – E2) (26) como se ilustra en la Figura 3. Esta estructura se localiza en la membrana mitocondrial interna y se encarga de catalizar la decarboxilación oxidativa de los sustratos de los ácidos cetónicos (27-29). Se desconoce la causa de la destrucción específica de esas células.
Consecuencias de la colestasis
Son múltiples las consecuencias y complicaciones relacionadas con la colestasis de la CBP (2-4). Entre estas se reconocen frecuentemente la fatiga, el prurito, la osteoporosis y osteopenia, la hiperlipidemia, la diarrea crónica y la malabsorción de vitaminas liposolubles. Las vitaminas comprometidas son la A que conlleva a alteraciones de la visión; la vitamina D asociada a osteopenia y alteraciones del metabolismo del calcio; deficiencia de vitamina E relacionada con neuropatía periférica y miopatía; y la vitamina K que causa alteración de los factores de coagulación dependientes (2).
Se ha encontrado que la entidad se asocia con enfermedades autoinmunes como el Síndrome de Sjögren, psoriasis, artritis reumatoidea y tiroiditis de Hashimoto, entre otras, que conllevan a complicaciones per se de cada entidad (30).
Por último, todas las consecuencias relacionadas con la hipertensión portal como son las várices esofágicas, ascitis, el síndrome hepatorrenal entre otros y en su estadio avanzando, el carcinoma hepatocelular.
Manifestaciones clínicas
Al momento del diagnóstico, el 60% de todos los pacientes pueden ser asintomáticos y conforme progresa la enfermedad, van apareciendo los síntomas (11). Después de 20 años, sólo el 5% permanece sin síntomas (31). Los síntomas más comunes son la fatiga y el prurito (2,32) como lo muestra la Tabla 1. La fatiga impacta negativamente el desempeño de las actividades físicas y en las esferas psicosocial y cognitiva, como se ha demostrado utilizando la escala Fatigue Impact Scale y el cuestionario PBC 40 (2).

El prurito, es un síntoma característico de la entidad. Ocurre en 20 a 70% de los pacientes, independiente del estadio y puede ser su primera manifestación (33). En el 75% de los pacientes precede el diagnóstico (34). Después de aparecer, su severidad es variable y puede disminuir en estados avanzados, conforme se deteriora la función sintética del hígado (33). El prurito es generalizado, más intenso y frecuente por las noches (33,34). Siempre necesita tratamiento para aliviarlo. A diferencia de otros pruritos, el rascado lo alivia muy poco (35) y por la intensidad de este se producen escoriaciones, foliculitis, liquenificación, aunque rara vez prurigo nodular (36).
Característicamente afecta las palmas de manos y plantas de los pies (34). El rascado crónico produce escoriaciones e hiperpigmentación de las zonas afectadas. Sin embargo, la parte media de la espalda al no poder ser alcanzada por la mano, se mantiene normal y contrasta con las zonas adyacentes, dando lo que se conoce como el "signo de la mariposa". El prurito puede ser severo, e impedir conciliar el sueño y en casos extremos producir depresión e incluso ideas suicidas (2). En ocasiones puede afectar tanto la calidad de vida de las personas que constituye una indicación para trasplante hepático. Al igual que la fatiga, cuando aparece al comienzo de la enfermedad, es un predictor de mal pronóstico y menor respuesta al UDCA (2-4).
La patogénesis del prurito es desconocida, en adición a ser un síntoma extremadamente subjetivo (37). Se ha sugerido que la colestasis, puede impedir la eliminación por la bilis de sustancias "pruritógenas", las cuales serían liberadas a la circulación sistémica y estimularía terminaciones nerviosas "pruritoceptivas" en la piel y de ahí se transmitiría el estímulo al cordón espinal y al cerebro (35). Diferentes "pruritógenos" han sido estudiados, incluyendo histamina, sustancia P, ácidos biliares y opioides endógenos como la encefalina, pero ninguno de esos ha sido demostrado de manera conclusiva. Más recientemente se ha encontrado que el ácido lisofosfatídico (ALSF) y la enzima autoxina pueden ser los más importantes (33). Se ignoran los mecanismos de producción de tales sustancias, pero se considera que factores retenidos en la bilis estimularían la transcripción de la autaxina y esta a su vez, aumentaría los niveles de ASLF, el cual es un potente activador neuronal que actúa directamente en las fibras nerviosas "pruritoceptivas" (38).
Otras alteraciones cutáneas incluyen hiperpigmentación y la aparición de xantelasmas y xantomas (2). Estos dos últimos, se deben al depósito de colesterol, debido a la acumulación al no poderse eliminar en las secreciones biliares (10). Conforme progresa la enfermedad, aparece ictericia.
Por la colestasis crónica, progresivamente, se deteriora la absorción de vitaminas liposolubles (A, D, E, K) con las manifestaciones características de sus deficiencias (2,3,7). En adición, se puede producir esteatorrea y mala-absorción (2,3).
Cuando se produce cirrosis aparecen las manifestaciones, que dependerán del grado de hipertensión portal e insuficiencia hepática que incluye ascitis, circulación colateral, arañas vasculares, esplenomegalia, encefalopatía, atrofia muscular, desnutrición, sangrado por várices, síndrome hepato- renal, e infección del líquido ascítico, entre otras (2,6).
Alteraciones bioquímicas. La CBP se caracteriza por aumento de la fosfatasa alcalina y de la γ-glutamiltransferasa (GGT) en el inicio de la entidad, incluso antes de la aparición de la sintomatología. Esta elevación es la marca característica de la colestasis y se correlaciona con la severidad de la inflamación y de la ductopenia (7) y el aumento de la mortalidad o necesidad de trasplante, véase más adelante.
La bilirrubina es usualmente normal en estadios tempranos (2). El aumento de la bilirrubina, a expensas de la fracción directa, se correlaciona con enfermedad avanzada (5,39). El aumento progresivo de las bilirrubinas séricas es el mejor predictor de mal pronóstico (5,39). Junto con los niveles elevados de fosfatasa alcalina, los niveles más altos de bilirrubina, se correlacionan con enfermedad progresiva y niveles más bajos, recíprocamente se correlacionan con sobrevida sin necesidad de trasplante hepático, como fue recientemente demostrado en un estudio internacional con más de 4800 pacientes estudiados (40). En ese estudio, se encontró que el 86% de los pacientes con bilirrubina total igual o menor a una vez el límite superior normal, estaban vivos a los 10 años de participar en el estudio, en contraste con 41% cuando el nivel de bilirrubina total era más de una vez el límite superior normal. El riesgo de trasplante o muerte fue 5,1 a 10,7 veces más cuando los niveles estaban altos comparados con quienes los tenían normales. Así mismo, cuando la fosfatasa alcalina era menor o igual a dos veces el límite superior normal, la sobrevida era de 84% versus 62% cuando mayor a dos veces el límite superior normal (40). Los pacientes con niveles elevados de fosfatasa alcalina tuvieron un riesgo de 2 a 2,5 veces de trasplante o muerte comparados con los que tenían niveles normales. Los niveles elevados de ambas igualmente aumentaban la mortalidad.
Hay incremento de globulinas y disminución de la síntesis de proteínas como albúmina, protrombina, y factores de coagulación a medida que progresa la enfermedad (5).
Las aminotransferasas pueden estar ligeramente elevadas, pero es una alteración inespecífica (2). Al igual que en otras entidades, su elevación refleja inflamación o alteración de la integridad de la membrana de los hepatocitos. Cuando hay cirrosis, está elevada la relación AST/ALT (7,10).
Hallazgos inmunológicos. En los pacientes con CBP, se han encontrado más de 60 auto anticuerpos, la mayoría de los cuales son inespecíficos y están básicamente relacionados con enfermedades autoinmunes asociadas (40). En contraste, los anticuerpos anti-mitocondriales (AMA) son altamente específicos de la entidad y están presentes en el 95% de los pacientes (2,5), siendo inusualmente negativos (CBP con AMA negativos). Se consideran positivos cuando su concentración por inmuno-fluorescencia indirecta (IFI) es de 1:40 o más. Los títulos de AMA no se correlacionan con la severidad de la CBP (6). Cuando son negativos, está indicada la biopsia hepática para el diagnóstico, como se discutirá en la segunda parte de esta revisión. Los pacientes con CBP AMA positivos, tienen similares características clínicas y bioquímicas que los AMA negativos, al igual que necesidad de trasplante hepático o sobrevida (11). Los AMA son considerados "la marca" de CBP. Muy rara vez se encuentran en personas sanas, su prevalencia es menor de 1% en la población general (41,42). El 10 a 19% de las personas con AMA positivos, pero sin alteraciones bioquímicas o clínicas, desarrollarán CBP (42). La positividad de esos anticuerpos es un marcador de enfermedad preclínica. Desde el momento de la detección de los AMA hasta la aparición de la enfermedad transcurren 6 años (1-19 años) (41-43). Los AMA son una familia de enzimas localizadas en la membrana interna de las mitocondrias, que tienen el complejo 2-oxo-acid deshidrogenasa, que incluye el PDC-E2. Tales enzimas catalizan la decarboxilación de cetoácidos (43). Existen nueve subtipos de AMAs (AMA1- AMA9) (6). Cuatro de ellos (2, 4,8 y 9) están asociados con CBP y los AMA2 son los más frecuentemente utilizados en la práctica diaria (6,7,41-43).
Otros auto-anticuerpos son los antinucleares (ANA), los cuales pertenecen a dos familias distintas (44). La primera son ANAS inespecíficos para CBP como el Ro/ SSa, la/SSB, Scl 70 y anti histonas (44). Se encuentran en 30-50% de los pacientes con CBP (5,6). La segunda familia son los anti Sp100 y anti-gp 210, los cuales son altamente específicos para CBP y son útiles cuando los AMA son negativos y se consideran "surrogate" o sustitutos de los AMA (40,41). Los anti Sp100 se encuentran en el 30% de los pacientes con CBP y los anti gp-210, en el 20 – 50%. Ambos se asocian con enfermedad progresiva y mal pronóstico (2,6,11). Antes de concluir que los AMA son negativos, es necesario investigarlos intensamente incluyendo pruebas inmunohistoquímicas sensibles, así como también repetir las pruebas (40-43). En esos casos está indicada la biopsia hepática. Los complejos antígeno – anticuerpo se asocian con elevación de diferentes clases de inmunoglobulinas, con predominio por la IgM (6), a menudo 6,27 ± 0,66 g/L (concentración normal 1-2 g/L) (5). El mecanismo de elevación aun no es claro y puede estar baja si los AMA son negativos (6,7).
Los anticuerpos anti-músculo liso (ASMA) contra proteínas contráctiles de la clase IgM, se identifican en 1/3 de los pacientes (5,41). Los anticuerpos anticentrómero pueden ser positivos en el 30% de los casos e indican pronóstico, y tienen relación con desarrollo de hipertensión portal y de cirrosis (6,41).
CONCLUSIONES
La colangitis biliar primaria cada día es más reconocida a nivel mundial, lo cual se correlaciona con el aumento de su incidencia en los países de altos ingresos; a pesar de lo anterior, en Colombia aún no se tiene un adecuado control epidemiológico, por lo que no hay cifras de los casos en nuestro país y probablemente es una condición sub diagnosticada que deteriora la calidad de vida de los pacientes afectados por la falta de tratamiento sintomático y dirigido a la enfermedad, el cual puede mejorar considerablemente el pronóstico de la misma.
La entidad se ha descrito como multifactorial, en donde factores genéticos, ambientales, infecciosos, en incluso la microbiota, pueden estar presentes. Su fisiopatología no está totalmente esclarecida, sin embargo, se conoce que la lesión final es una destrucción lenta y progresiva de las células epiteliales biliares que conduce a una sintomatología inespecífica al inicio de su curso natural, como el prurito y la fatiga, hasta manifestaciones sistémicas importantes secundarias a la etapa final de cirrosis hepática.
Conflictos de intereses: Los autores declaran no tener ningún conflicto de interés.
Fuente de financiamiento: Ninguna.
REFERENCIAS BIBLIOGRÁFICAS
1. Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DEJ, Lindor K, et al. Changing nomenclature for PBC: From "cirrhosis" to "cholangitis". Dig Liver Dis. 2015;47(11):924-6. [ Links ]
2. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386(10003):1565-75. [ Links ]
3. Kouroumalis E, Notas G. Primary biliary cirrhosis: From bench to bedside. World J Gastrointest Pharmacol Ther. 2015;6(3):32-58. [ Links ]
4. Rose NR, Bona C. Defining criteria for autoimmune diseases (Witebsky’s postulates revisited). Immunol Today. 1993;14(9):426-30.
5. Reshetnyak VI. Primary biliary cirrhosis: Clinical and laboratory criteria for its diagnosis. World J Gastroenterol. 2015;21(25):7683-08. [ Links ]
6. Yamagiwa S, Kamimura H, Takamura M, Aoyagi Y. Autoantibodies in primary biliary cirrhosis: Recent progress in research on the pathogenetic and clinical significance. World J Gastroenterol. 2014;20(10):2606-12. [ Links ]
7. Corrigan M, Hirschfield GM. Aspects of the pathophysiology of primary biliary cirrhosis. Dig Dis. 2015;33(2):102-8. [ Links ]
8. Flores A, Mayo MJ. Primary biliary cirrhosis in 2014. Curr Opin Gastroenterol 2014;30(3):245-52. [ Links ]
9. Poupon R. Non-invasive assessment of liver fibrosis progression and prognosis in primary biliary cholangitis. Dig Dis. 2015;33 Suppl 2(suppl 2):115-7.
10. Addison T, Gull W. On a certain affection of the skin vitiligo idea-α-plana β-tuberosa. Guys Hosp Rep. 1851;7:265-77. [ Links ]
11. Purohit T, Cappell MS. Primary biliary cirrhosis: Pathophysiology, clinical presentation and therapy. World J Hepatol. 2015;7(7):926-41. [ Links ]
12. Trivedi PJ, Hirschfield GM. Primary biliary cirrhosis: renaming primary biliary cirrhosis-clarity or confusion? Nat Rev Gastroenterol Hepatol. 2015,12(12):678-79.
13. Herrera PY, Restrepo JC, Duque SH, Arango GC. Reporte de casos: Reporte de dos casos de síndrome de sobreposición entre hepatitis autoinmune y cirrosis biliar primaria. Rev Col Gastroenterol. 2010;25(4):399-04. [ Links ]
14. Talwalkar JA, Lindor KD. Primary biliary cirrhosis. Lancet. 2003;362(9377):53-61. [ Links ]
15. Marzorati S, Lleo A, Carbone M, Gershwin ME, Invernizzi P. The epigenetics of PBC: The link between genetic susceptibility and environment. Clin Res Hepatol Gastroenterol. 2016;40(6):650-9. [ Links ]
16. Mattner J. Impact of microbes on the pathogenesis of primary biliary (PBC) and primary sclerosing cholantitis (PSC). Int J Mol Sci. 2016;17(11):E1864. [ Links ]
17. Schrumpf E, Kummen M, Valestrand L, Greiner TU, Holm K, Arulampalam V, et al. The gut microbiota contributes to a mouse model of spontaneous bile duct inflammation. J Hepatol. 2017;66(2):382-9. [ Links ]
18. Verdier J, Luedde T, Sellge G. Biliary mucosal barrier and microbiome. Viszeralmedizin. 2015;31(3):156-61. [ Links ]
19. Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview based study of 1032 patients. Hepatology. 2005;42(5):1194-202. [ Links ]
20. Juran BD, Lazaridis KN. Environmental factors in primary biliary cirrhosis. Semin Liver Dis. 2014;34(3):265-72. [ Links ]
21. Prince MI, Ducker SJ, James OF. Case-control studies of risk factors for primary biliary cirrhosis in two United Kingdom populations. Gut. 2010;59(4):508-12. [ Links ]
22. Eckburg PB, Bik EM, Bernstein, CN, Purdom M, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635-8. [ Links ]
23. Hofmann AF. Biliary secretion and excretion in health and disease: current concepts. Ann. Hepatol. 2007;6(1):15-27. [ Links ]
24. Lee, JY, Arai H, Nakamura Y, Fukiya S, Wada M, Yokota A. Contribution of the 7β-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J Lipid Res. 2013;54(11):3062-9. [ Links ]
25. Islam KB, Fukiya S, Hagio M, Fuji N, Ishizuka S, Ooka T, et al. Bile Acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141(5):1773-81. [ Links ]
26. Sharon D, Mason AL. Role of novel retroviruses in chronic liver disease: assessing the link of human betaretrovirus with primary biliary cirrhosis. Curr Infect Dis Rep. 2015;17(2):460. [ Links ]
27. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ. Primary biliary cirrhosis. Hepatology. 2009;50(1):291-308. [ Links ]
28. Cha S, Leung PS, Gershwin ME, Fletcher MP, Ansari AA, Coppel RL. Combinatorial autoantibodies to dihydrolipoamide acetyltransferase, the major autoantigen of primary biliary cirrhosis. Proc Natl Acad Sci U S A. 1993;90(6):2527-31. [ Links ]
29. Mutimer DJ, Fussey SP, Yeaman SJ, Kelly PJ, James OF, Bassendine MF. Frequency of IgG and IgM autoantibodies to four specific M2 mitochondrial autoantigens in primary biliary cirrhosis. Hepatology. 1989;10(4):403-7. [ Links ]
30. Floreani A, Franceschet I, Cazzagon N, Spinazzè A, Buja A, Furlan P, et al. Extrahepatic autoimmune conditions associated with primary biliary cirrhosis. Clin Rev Allergy Immunol. 2015;48(2-3):192-7. [ Links ]
31. Prince M, Chetwynd A, Newman W, Metcalf JV, James OF. Survival and symptom progression in a geographically based cohort of patients with primary biliary cirrhosis: follow-up for up to 28 years. Gastroenterology. 2002;123(4):1044-51. [ Links ]
32. Lindor K. Ursodeoxycholic acid for the treatment of primary biliary cirrhosis. N Engl J Med. 2007;357:1524-29. [ Links ]
33. Hegade VS, Bolier R, Oude Elfering RP, Beuers U, Kendrick S, Jones DE. A Systematic approach to the management of cholestatic pruritus in primary biliary cirrhosis. Frontline Gastroenterol. 2016;7(3):158-66. [ Links ]
34. Rische E, Azarm A, Bergasa NV. Itch in primary biliary cirrhosis: a patient`s perspective. Acta Derm Vebereol. 2008;88(1):34-7. [ Links ]
35. Hegade VS, Kendrick SF, Jones DE. Drug treatment of pruritus in liver diseases. Clin Med (Lond). 2015;15(4):351-7. [ Links ]
36. Kremer AE, Namer B, Bolier R, Fischer MJ, Oude Elferink RP, Beuers U. Pathogenesis and management of pruritus in PBC and PSC. Dig Dis. 2015;33(suppl 2):164-75. [ Links ]
37. Quarmeti C, Muratori P, Lalanne C, Fabbbri A, Menichella R, Granito A, et al. Fatigue and pruritus at onset identify a more aggressive subset of primary biliary cirrhosis. Liv Intern. 2015;3(2)5:636-41. [ Links ]
38. Kremer AE, Oude Elferink RP, Beuers U. Pathophysiology and current management of pruritus in liver disease. Clin Res Hepatol Gastroenterol. 2011;35(2):89-97. [ Links ]
39. Trivedi PJ, Corpechot C, Pares A, Hirschfield GM. Risk stratification in autoimmune cholestatic liver diseases: Opportunities for clinicians and trialists. Hepatology. 2016;63(2):644-59. [ Links ]
40. Lammers WJ, van Buuren HR, Hirschfield GD, Janssen HLA, Invernizzi P, Mason AL, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow study. Gastroenterology. 2014;147(6):1338-49. [ Links ]
41. Nakamura M. Clinical significance of autoantibodies in primary biliary cirrhosis. Semin Liver Dis. 2014;1(212):334-40. [ Links ]
42. Yamagiwa S, Kamimura H, Takamura M, Aoyagi Y. Autoantibodies in primary biliary cirrhosis: recent progress in research on the pathogenetic and clinical significance. World J Gastroenterol. 2014;20(10):2606-12. [ Links ]
43. Imam MH, Lindor KD. The natural history of primary biliary cirrhosis. Sem Liv Dis. 2014;34(3):329-33. [ Links ]
44. Vergani D. Towards the serological diagnosis of primary biliary cirrhosis. Liv Int. 2015;35(2):299-301. [ Links ]
Correspondencia:
Wiliam Otero Regino
E-mail: waoteror@gmail.com
Recibido: 11-4-2017
Aprobado: 30-8-2017

