











 texto en
texto en  Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Las ictiosis congénitas autosómicas recesivas son un grupo de trastornos cutáneos de origen genético y fenotipos heterogéneos. Estas incluyen ictiosis arlequín, ictiosis laminar y eritrodermia ictiosiforme congénita bullosa1). La ictiosis arlequín, también conocida como queratosis difusa fetal, es un trastorno grave, muy poco presente y generalmente mortal2). Su incidencia es de 1 por cada 300,000 nacidos vivos. Su patrón de herencia es autosómico recesivo por la mutación del gen de la proteína de unión a trifosfato de adenosina A12 (ABCA12), que provoca alteraciones del transporte de lípidos en la epidermis(3,4).
La ictiosis arlequín es una afección con apariencia clínica peculiar, ya que la alteración cutánea y la acumulación de escamas producen placas hiperqueratósicas generalizadas, de color blanquecino o marrón oscuro, con fisuras profundas en casi todo el cuerpo. Otras características incluyen ectropión, boca en forma de pez y contracturas de los miembros(1). La ecografía y el análisis genético son herramientas útiles para el diagnóstico prenatal3). Se presenta un caso de diagnóstico prenatal de ictiosis arlequín.
COMUNICACIÓN DEL CASO
Una paciente de 18 años, primigesta, fue referida a la consulta de alto riesgo prenatal por hallazgos faciales fetales anormales en la ecografía fetal de rutina a las 28 semanas. La paciente acudió regularmente a la consulta prenatal de su centro y el embarazo había transcurrido sin complicaciones. La ecografía rutinaria a las 12 y 20 semanas no mostró alteraciones fetales. Los resultados de las pruebas prenatales no invasivas para síndrome de Down, defectos del tubo neural y trisomía 18 fueron negativos. Los padres tenían relación de parentesco lejana y negaban antecedentes de trastornos familiares genéticos, cutáneos hereditarios o contacto con pesticidas y sustancias radiactivas.
La ecografía estándar mostró embarazo intrauterino con feto masculino en presentación de nalgas y biometría correspondiente a 24 semanas. El volumen de líquido amniótico estaba disminuido para la edad gestacional (por debajo del percentil 10) con aspecto ecogénico y turbio, con partículas flotantes densas. Los rasgos faciales fetales incluían labio superior e inferior marcadamente engrosados y en forma similar a la boca de pez, eversión bilateral de los párpados (ectropión), hueso nasal corto con nariz chata, orejas deformadas y catarata congénita. Además, también fue observada columna deformada, anomalías de las extremidades con dedos de manos y pies hipoplásicos e incurvados en flexión (figura 1). Los movimientos fetales eran escasos. Las imágenes de ecografía tridimensional confirmaron los hallazgos anormales (figura 2). El diagnóstico prenatal sugería la posibilidad de ictiosis de arlequín. Los padres dieron permiso para realizar la amniocentesis para evaluar el cariotipo fetal, el cual no mostró anomalías cromosómicas.
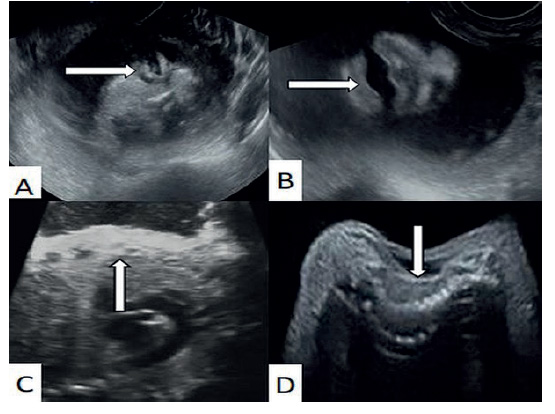
Figura 1 Imagen ecográfica de la ictiosis arlequín. La flecha señala A) Ectropión, B) Boca en forma de O, C) Piel engrosada y D) deformidad de la columna vertebral.
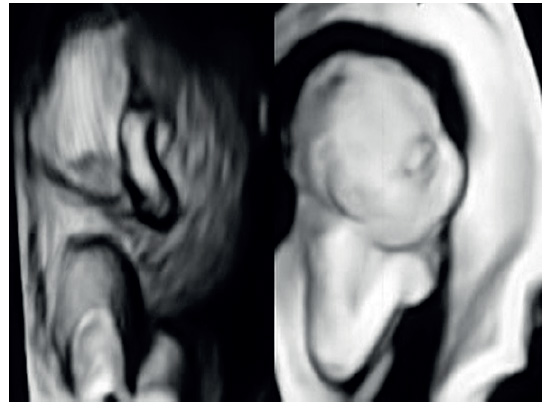
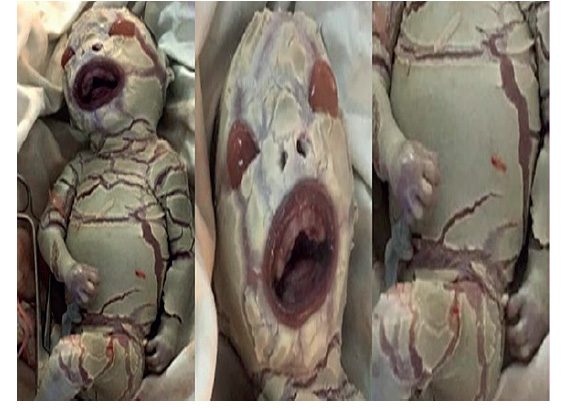
A las 30 semanas, la paciente asistió a la emergencia obstétrica por contracciones uterinas, hemorragia vaginal y disminución de los movimientos fetales. La evaluación ecográfica mostró ausencia de frecuencia cardiaca fetal, motivo por el cual se decidió la evacuación uterina. Se obtuvo mortinato de 1,300 gramos y 25 centímetros de longitud. Al examen físico se observó que la piel estaba seca y engrosada (consistente con hiperqueratosis), dividida en forma irregular por fisuras eritematosas profundas entre placas gruesas, amarillentas, parecidas a escamas gruesas que afectaban toda la superficie corporal. También se observó eversión de párpados superiores e inferiores; boca de pez, nariz plana ensanchada y orejas anormalmente aplanadas con lóbulos fusionados. Las extremidades estaban en posición semiflexionada persistente, con contracturas en los dedos de manos y los pies incurvados (figura 3).
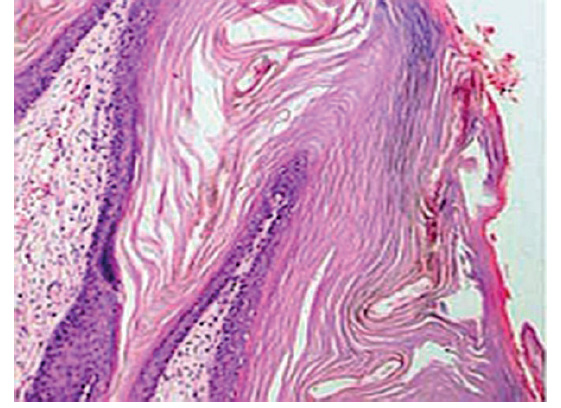
Se tomó una muestra de piel del tórax que fue enviada a anatomía patológica. La evaluación microscópica mostró estrato córneo extremadamente grueso, indicativo de hiperqueratosis, con escasas células musculares esqueléticas en dermis y tejido celular subcutáneo (figura 4). La secuenciación de ADN reveló la variante homocigota de ABCA12 indicativa de ictiosis congénita autosómica recesiva. Todos los hallazgos eran compatibles con el diagnóstico de ictiosis arlequín.
DISCUSIÓN
La ictiosis arlequín es una forma de genodermatosis severa caracterizada por hiperqueratosis y descamación de la epidermis5). Su incidencia estimada es de 1 caso por cada 300,000 nacimientos, sin predisposición sexual, con patrón de herencia autosómico recesivo y tasa de recurrencia en embarazos posteriores del 25%6). La ictiosis arlequín es causada por la mutación del gen ABCA12 que se encuentra en el cromosoma 2 (2q34). Esta alteración de la función provoca cambios en los depósitos de lípidos en el estrato córneo, lo que conduce a alteración de la barrera cutánea e hiperqueratinización compensatoria(7). La formación de las capas lipídicas intercelulares en los queratinocitos epidérmicos es esencial para la función de barrera epidérmica y la formación defectuosa de las capas lipídicas produce pérdida de la función de barrera epidérmica e hiperqueratosis anormal, provocando la aparición de las lesiones cutáneas características(2,3).
El diagnóstico prenatal de la ictiosis arlequín es esencial para futuros embarazos2). Las características ecográficas fundamentales son presencia de ectropión, pabellón auricular anormal, nariz plana, piel engrosada con apariencia de armadura, labios engrosados con estado sostenido de boca abierta (boca de pez), fijación en flexión de las extremidades, disminución o ausencia de movimientos fetales y partículas gruesas flotando en el líquido amniótico (signo del copo de nieve)(6). Por lo general, el diagnóstico ecográfico puede realizarse con certeza en el tercer trimestre, debido a que la queratinización cutánea comienza a las 22 a 24 semanas de gestación. La ecografía tridimensional contribuye a la evaluación de la morfología facial. Sin embargo, no sustituye a la ecografía bidimensional(3).
Los análisis de secuenciación del ADN en los casos de ictiosis arlequín son fiables y concluyentes(4). El diagnóstico está basado en identificar las mutaciones de ABCA121). La proteína es miembro de la superfamilia de transportadores dependientes de ATP, que se unen e hidrolizan a diversas moléculas a través de membranas o vesículas(8). Esta proteína está presente en gránulos laminares de los queratinocitos que regulan el tráfico de lípidos9). El diagnóstico puede confirmarse mediante muestras de vellosidades coriónicas o de líquido amniótico10). En pacientes con antecedentes familiares, la fetoscopia con biopsia de piel y el examen ultraestructural de las células del líquido amniótico pueden ser útiles, aunque no es recomendado en la actualidad por los avances en pruebas no invasivas como la ecografía(11).
El principal diagnóstico diferencial de la ictiosis arlequín en el periodo prenatal es el síndrome de Neu-Laxova. La boca de pez, ausencia de microcefalia y la falta de edema son típicos de la ictiosis arlequín, mientras que las cataratas y el cordón umbilical corto son característicos del síndrome de Neu-Laxova(11). Por otra parte, la ictiosis arlequín fetal pueden confundirse con la macroglosia fetal y el angioma fetal congénito de tipo tumoral12). En ambas condiciones, la lengua extendida fuera de la boca hace que esta quede fija en forma de O. Sin embargo, la macroglosia siempre está asociada a trastornos genéticos como síndrome de Down y de Beckwith-Wiedemann13). La lengua engrosada en el hemangioma congénito fetal suele mostrar aumento del flujo sanguíneo en el Doppler color14).
Los diagnósticos diferenciales en neonatos incluyen artrogriposis, aplasia cutis, enfermedad de Gaucher, síndrome de Sjogren-Larsson, síndrome de Conradi-Hunermann-Happle y tricotiodistrofia(15). Los casos graves pueden ser diagnosticados fácilmente de forma errónea como epidermólisis bullosa o ictiosis sindrómica9).
Aquellos neonatos con ictiosis arlequín tienen un aspecto clínico distintivo, con piel engrosada, de color blanquecino a amarillento, como una armadura, con fisuras que dividen la piel en secciones poligonales o en forma de diamante y grietas rojizas en todo el cuerpo7). Esto acompañado de ectropión y boca redonda y persistentemente abierta. No existe tratamiento definitivo para esta condición. El pronóstico es desfavorable y la probabilidad de muerte neonatal es elevada, a pesar del tratamiento médico de apoyo. La mayoría de las muertes ocurren poco después del parto debido a infecciones, pérdida de calor, deshidratación, alteraciones electrolíticas o dificultad respiratoria3).
En conclusión, la ictiosis arlequín es un trastorno genético autosómico recesivo, poco frecuente y generalmente mortal en los primeros días de vida. El diagnóstico genético prenatal debe aconsejarse a las parejas con niños previamente afectados. Los rasgos característicos en la ecografía prenatal tienden a aparecer en forma tardía, por lo que las evaluaciones deben repetirse en el tercer trimestre, incluso cuando la exploración anatómica del segundo trimestre sea normal.

