











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Se calcula que existen entre 15 a 20 millones de infectados por el virus de la hepatitis delta (VHD) a nivel mundial, lo cual corresponde al 5% de los infectados por el virus de hepatitis B (VHB) 1. Esta infección puede ocurrir como una superinfección o una coinfección VHB/ VHD. La infección crónica VHB/VHD puede ocasionar formas más severas de la enfermedad con una mayor progresión a fibrosis del hígado y carcinoma hepatocelular (HCC) 2.
El VHD es un virus ARN de cadena simple circular covalentemente cerrado. Es defectuoso porque requiere del antígeno de superficie (HBsAg) del virus de hepatitis B (VHB) para infectar y replicarse en los hepatocitos. Este virus tiene entre 1679 a 1697 nucleótidos, con un único marco abierto de lectura (siglas ORF del inglés open reading frame) que codifica dos isoformas de la proteína Antígeno Delta (HDAg): HDAg-L y HDAg-S 3, los cuales se diferencian en los 19 aminoácidos (aac.) adicionales en el extremo Carboxilo- terminal del HDAg 4. La isoforma HDAg-S promueve la replicación viral y la HDAg-L participa en el ensamblaje del virión 5.
El VHD se clasifica en 8 genotipos cuya divergencia intergenotípica está entre el 35% al 40% mientras que la heterogeneidad intragenotípica es <20%. La distribución geográfica de estos genotipos es variada: el VHD-1 tiene distribución mundial y los otros genotipos son específicos de una región geográfica, los VHD-2 y VHD-4 son prevalentes en Japón (Asia), el VHD-3 es prevalente en Latinoamérica, principalmente en la Amazonía y en poblaciones amerindias; y los VHD-5 al VHD-8 en África 6.
La prevalencia de la infección por VHD es variada según zona geográfica. África, Mediterráneo, Asia, Japón, Taiwán, Pakistán, Medio Oriente y Sudamérica son consideradas como áreas de mayor prevalencia. Sudamérica, en las últimas 3 décadas, ha reportado una prevalencia del 22,37%, sin embargo, hay que tener en cuenta que la mayoría de los estudios han sido realizados en zonas consideradas endémicas y con aislamiento geográfico 7.
En el Perú hay algunos estudios de la prevalencia del VHD los cuales indican como zonas de mayor prevalencia a las comunidades nativas de la Amazonía y las áreas andinas de Abancay, Ayacucho y Andahuaylas. Un estudio en 8 localidades del río Pampas (Ayacucho-Andahuaylas), en escolares HBsAg positivos, determinó una prevalencia del 16,7% para VHD 8; mientras que en la ciudad Huanta, el 14,7% de escolares anti HBc positivos tuvieron infección por el VHD 9. En pobladores amazónicos con HBsAg positivos la prevalencia fue 39% 10 y en pobladores de Abancay con infección a VHB fue del 9% 11. Entre 1992-1993, en un brote en soldados asentados en la selva amazónica del Perú con diagnóstico clínico de VHB, se determinó la presencia del VHD en el 64% de ellos; asimismo, se determinó que correspondían al genotipo 312. El VHD-3, reportado en Perú y que circula en la Amazonía de Latinoamérica se asocia con un mayor riesgo de insuficiencia hepática aguda y con brotes 1,2, por ello, el conocimiento de la circulación del VHD-3 es de importancia.
Estudios actuales indican que la prevalencia del VHD se ha reducido en los últimos años, pasando de una zona de alta endemicidad a una zona de baja endemicidad. Las campañas de vacunación contra VHB realizadas por el MINSA y el tamizaje para VHB han contribuido en disminuir la prevalencia del VHD en las regiones de mayor endemicidad.
Este estudio ha sido realizado para identificar el genotipo del VHD que se encuentra circulando en comunidades andinas como Huanta (Ayacucho) y en pueblos indígenas amazónicos: Kandozi, Chapra y Matsés (Loreto). Se ha estimado el rango de sustitución nucleotídica, la dinámica poblacional y el tiempo del ancestro común más reciente (tMRCA). Este estudio se justifica por la asociación que tiene el genotipo del VHD con las manifestaciones clínicas, la progresión de la enfermedad, el comportamiento biológico y la respuesta ante el tratamiento antiviral.
MÉTODOS
Población y tamaño muestral
Se realizó un estudio observacional, analítico y de corte transversal, se utilizaron 582 muestras provenientes de estudios previos realizados en las comunidades de Huanta, Matsés, Kandozi y Chapra que fueron recolectadas durante los años 2010 al 2012 y almacenadas en congelación en el Laboratorio de Hepatitis del Instituto Nacional de Salud (Lima, Perú). Estas muestras tenían resultado positivos al anti-HBc del VHB mediante el método ELISA. La muestra de cada estudio fue representativa de cada comunidad.
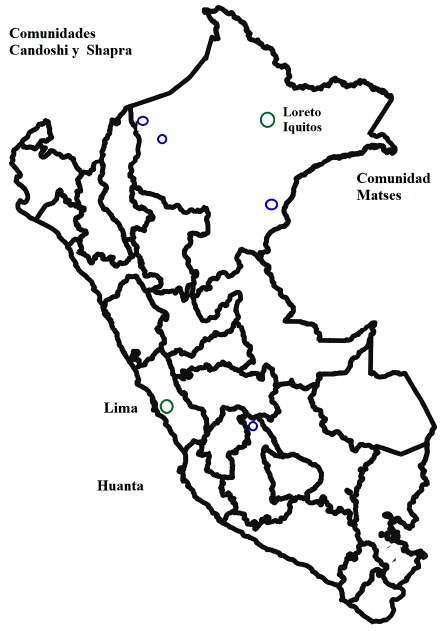
Las tres comunidades nativas amazónicas se encuentran ubicados en el departamento de Loreto. Los Kandozi y Chapra están localizados en la provincia de Datem del Marañón, en la cuenca de Pastaza y Morona. Los Matsés se ubican en la provincia de Requena, en los márgenes de los ríos Gálvez y Yaquirana y la quebrada Añushiyacu. La provincia de Huanta, capital de la región Ayacucho, es un valle interandino ubicado en la vertiente oriental de la cordillera de los Andes en Perú (Figura 1).
Análisis de laboratorio
Serología y carga viral
En las instalaciones del Laboratorio de Hepatitis, durante el año 2017, se realizaron los análisis de laboratorio para la determinación de los marcadores serológicos IgG o IgM del VHD utilizando pruebas ELISA de la marca Wantai (Beijing wantai Biological Pharmacy) y el lavador y lector de ELISA marca Robonik. Todos los análisis fueron realizados siguiendo las indicaciones del fabricante.
Para las muestras de portadores crónicos del VHB, se determinó la carga viral del VHB por reacción cuantitativa en cadena de la polimerasa en tiempo real (qPCR) utilizando los kits COBAS AmpliPrep /COBAS TaqMan VHB Test, versión 2.0 (Roche Molecular Diagnostics, Branchburg, Nueva Jersey), cuyo límite inferior de detección es de 20 UI/mL.
Extracción del ARN y nRT-PCR del VHD
A las muestras positivas al IgG o IgM del VHD se les extrajo el ARN utilizando el kit comercial QIAamp viral RNA (Qiagen, Germany) a partir de 140 µl de suero. El ARN extraído fue denaturado a 95°C por 5 minutos junto con los oligonucleótidos aleatorios, se agregaron 12 µL del denaturado a la mezcla de transcripción reversa para la síntesis de cDNA (RT-PCR) empleándose el kit High Capacity cDNA Reverse Transcription (Applied Biosystems-Life Technologies, Carlsbad, CA). 10 µL cDNA, con 40 µL de mezcla de reacción [buffer 1x, 1,5 mM MgCl2, 0,2 de cada dNTPs, 0,5 pmol/µL de cada oligonucleótido 853IU-1302OD, y 0.025 U/ µL de Taq DNA polimerase (Invitrogen). Para el PCR anidado (nRT-PCR-VHD) se utilizó 5 µl del primer amplificado y 45 µl de la mezcla de PCR [buffer 1x, 1,5 mM MgCl2, 0,25 de cada dNTPs, 0,5 pmol/µL de cada oligonucleótido (HDV-E y HDVA) y 0,025 U/µL de Taq DNA polimerase (Invitrogen). Las condiciones del PCR fueron: 94°C por 2 m, seguido por 40 ciclos de 30s a 94°C, 50s a 58°C y 45s a 72°C con una extensión final de 5 min a 72°C. Los productos de PCR fueron visualizados en gel de agarosa al 1,5%, se obtuvieron amplificados de 403 pb y 374 pb respectivamente 13,14.
Secuenciación del VHD
Los productos del PCR amplificados fueron purificados con el kit QIAquick Gel Extraction (QIAGEN, Hilden, Germany). Se cuantificó el producto utilizando el nanodrop. El secuenciamiento fue realizado con los oligonucleótidos anteriormente descritos y el kit Big Dye Terminator v3.1 Cycle Sequencing (Applied Biosystems) en el analizador genético automatizado ABI 3500 (Applied Biosystem, Foster city, CA, USA).
Análisis filogenético
El análisis filogenético fue realizado con 111 secuencias de referencia de la base de datos del Genbank (National Center for Biotechnology Information, Bethesda, MD, USA), correspondientes a los genotipos 1 al 8 del VHD. Las 42 secuencias peruanas del estudio fueron editadas utilizando el programa Chromas 2.6.5. Para elaborar las secuencias consenso se utilizó el programa Bioedit v7.2. El alineamiento de todas las secuencias fue realizado con el ClustalW v.1.6 del programa Mega V7.0 (https://www.megasoftware.net/), el corte se realizó con el software Gblocks server.
El análisis molecular bayesiano de las secuencias fue realizado utilizando el Markov Chain Monte Carlo (MCMC) del programa Beast V2.5.2, con el modelo de sustitución nucleotídica GTR+G (General Time Reversible, gamma distributed) obtenido con el Jmodeltest v2.1.10. Para la medida del tiempo filogenético se utilizaron dos modelos de relojes: relaxed uncorrelated lognormal molecular clock y el relaxed uncorrelated Exponential molecular clock, con 30 millones de sustituciones. Se escogió el mejor reloj molecular por Comparación del Factor Bayes (BF). Se utilizó el Tree Annotator para obtener el árbol con clado de máxima credibilidad (maximum clade credibility - MCC) a partir de la evaluación de cada clado de los 30000 árboles con exclusión de 10% del burn-in. El figtree v1.4.4 fue utilizado para visualizar el árbol filogenético. También se utilizaron los programas Beuti y Tracer v1.7.
Análisis evolutivo
Para el análisis evolutivo se utilizaron 47 secuencias de referencia del GenBank que correspondían al VHD-3 de Sud América provenientes de Brasil, Venezuela, Colombia, Bolivia y Perú, y las 42 secuencias de este estudio. Se utilizó el software Beast V2.5.2 con el modelo Bayesiano Coalescente Exponential Population y el reloj relaxed uncorrelated Exponential molecular clock, con 50 millones de sustituciones y un burn-in del 10%. El modelo de sustitución nucleotídica obtenido en el Jmodeltest fue el HKY+G. Se evaluó la convergencia de las cadenas con el software Tracer v1.7, se reconstruyó la dinámica poblacional en el tiempo con el modelo paramétrico Exponential Group Rate del Coalecente Demografic Reconstruction y con un intervalo de alta densidad probabilística (HPD) al 95%.
Análisis estadístico
La información demográfica y virológica de cada muestra del estudio fue ingresada a una base de datos anónima en Excel 2013. Se realizó un análisis descriptivo mediante frecuencias absolutas y relativas de variables categóricas. Los análisis estadísticos se realizaron en el programa STATA v.14.0, (College Station, Texas) para Windows.
RESULTADOS
Datos demográficos y virológicos
De las 582 muestras previamente reportadas como positivas a Anti-HBc por el método ELISA, el 22,3% (n = 130) correspondían a muestras de pobladores de la provincia de Huanta, el 13,4% (n = 78) a la etnia Kandozi, el 6,2% (n = 36) a la etnia Chapra y el 58,1% (n = 338) a la etnia Matsés.
Las mujeres representaron el 52,8% (n = 307). La media de la edad fue de 38 años (mínimo de y máximo de 86). Los menores de 5 años representaron un 0,7% (4/544) y los menores de 10 años el 2,8% (15/544). El 17,4% (101/582) presentan coinfección VHB-VHD, el 17,4% tuvieron ELISA IgG-VHD positivos y el 9,1% tuvieron ELISA IgM-VHD positivo. El 12,7% (74/582) fueron HBsAg positivo. Los valores de la carga viral para el VHB fueron: no detectables en una muestra, < 2000 UI/ mL en 37 muestras y >2000 UI/mL en 4 muestras. En cuanto a la procedencia, el 4,9% (4/101) fueron de Huanta, el 43,6% (44/101) de Kandozi, el 31,7% (32/101) de Matsés y el 19,8% (20/101) de Chapra.
El 53,5% (54/101) fueron positivos al nRT-PCR-VHD siendo el fragmento amplificado correspondiente a los nucleótidos 857 al 1322, los cuales permitieron identificar el genotipo del VHD. De las 50 secuencias amplificadas se consideraron 42 secuencias para el análisis filogenético y evolutivo. 8 secuencias no fueron de buena calidad. (Tabla 1).
Tabla 1. Prevalencia de IgG-VHD y IgM-VHD en las comunidades de la Amazonía (Matsés, Kandozi y Chapra) y el valle interandino de Huanta en pacientes anti HBc reactivos.
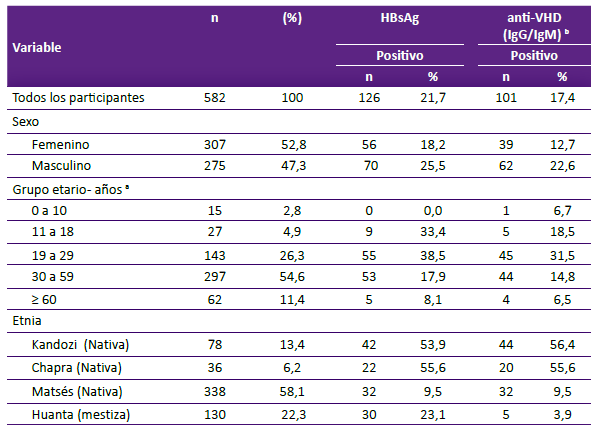
a El reporte del grupo etario comprende a 544 muestras.
b El reporte del grupo etario comprende a 99 muestras (Coinfección VHB-VHD)
Análisis nucleotídico y aminoacídico
Las secuencias obtenidas correspondieron al segmento que codifican los aac 93 al 214 de la proteína HDAg y a una región no traducible en el extremo C- terminal. El porcentaje de bases Guanina y Citosina fue del 62%. El porcentaje de similaridad entre las secuencias del estudio con la secuencia peruana reportada en 1993 fue de 94,0% a 99,7%.
Se identificaron dos de los dominios de unión al ARN (RDB). El primer dominio está ubicado entre los aac 96 -106 (DQERRDHRRRK) y presenta un cambio en el aac R100Q. El segundo dominio está entre los aac 135 -145 (DDDERERRTAG) y presenta cambios en los aac D135E, D137E y T143A en algunas de las secuencias analizadas. Seguido a los dos dominios RDB, hay una región rica en Prolina y Glicina. En el extremo C-terminal del HDAg-L se ubica la señal de ensamblaje del virus (VAS), que corresponde a los aac 196 - 214 (YGFTPPPPGYYWVPG-CTQQ), en algunas de las secuencias analizadas se observó un cambio en los aac F198L, T199S y Y205H. La señal de prenilación (Py), la composición de aac CTQQ y la cola de poly(A) (952-TTTATT-957) no presentan variación en ninguna de las secuencias peruanas.
El blanco de la edición del ARN (nt1014) en la mayoría de las secuencias peruanas presenta timina (T), sólo 2 secuencias de Chapra y 1 de Kandozi presentan 1014C. El sitio de clivaje del ARN para el antigenoma se ubica en las posiciones 904 y 905 (CG) y es constante en las secuencias peruanas obtenidas en el estudio y en las secuencias de referencia utilizadas en el análisis.
Análisis filogenético y evolutivo
El análisis filogenético de las 42 secuencias obtenidas en este estudio y las 111 secuencias del VHD reportadas en el GenBank mostró que las secuencias de las comunidades Matsés, Kandoshi, Shapra y Huanta se agrupan en un clúster monofilético con las secuencias que corresponde al genotipo 3 que es el genotipo más prevalente en Sudamérica (Figura 2).
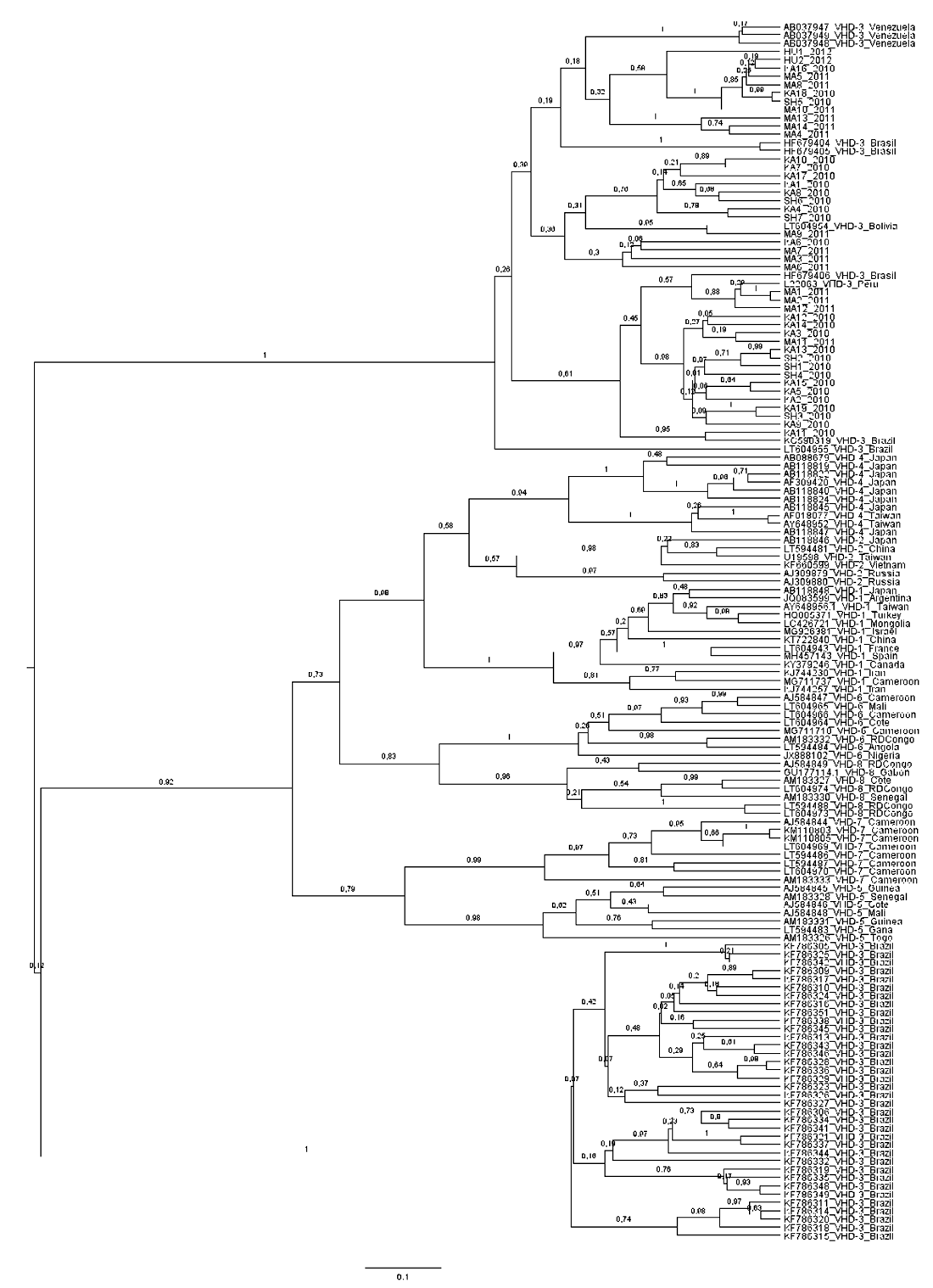
Figura 2. Árbol filogenético no enraizado del VHD fue estimado mediante el análisis bayesiano de 153 secuencias de hepatitis delta. Están incluidas las 42 secuencias obtenidas en este estudio. Las secuencias del genbank están identificadas por su número de accesión y el origen geográfico.
El análisis evolutivo mediante el modelo bayesiano Coalescente Exponential Population, para las 42 secuencias del estudio y las 47 secuencias del VHD-3 reportadas en el GenBank determinó que el rango de sustitución de nucleotídica del VHD-3 es de 2,047 x 10-3 sustituciones/ sitio/año. El valor estimado del ancestro común más reciente (tMRCA) para el VHD-3 que ha estado circulando en Sudamérica es de 1930 (HPD 95%) (Figura 3).
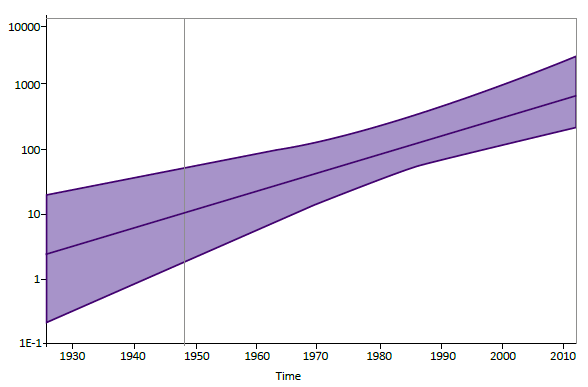
Figura 3. Dinámica poblacional de la diversidad genética del VHD-3. El eje (y) representa la diversidad genética relativa y el eje (x) el tiempo cronológico en años. La línea gruesa en azul indica la mediana y las líneas delgadas los intervalos de alta densidad probabilística (HPD) al 95% del Ne.r. Para la estimación se utilizaron 89 secuencias del VHD-3 de 321 nt.
DISCUSIÓN
En nuestro estudio encontramos la presencia del VHD en una región andina (Huanta) y en las comunidades amazónicas Matsés, Kandozi y Chapra. Estudios previos en el Perú reportan la presencia del VHD en algunas zonas geográficas, tales como Huanta, donde se encontró una prevalencia de 17,9% en portadores del VHB 9, mientras que en Abancay la prevalencia fue de 9% 11. En Andahuaylas y en selva amazónica la prevalencia del VHD en portadores del HBsAg fue de 16,7% 8) y 39%, respectivamente 10. En otras comunidades amazónicas como los Jíbaro, Pano y Arawak se encontró una prevalencia de 6,1%.
El alto porcentaje de muestras con valores de carga viral del VHB menores a 2000 UI/mL, estaría en concordancia con estudios que sugieren que el VHD suprime la replicación del VHB, produciendo carga viral VHB baja o por debajo del intervalo de confianza del método 15,16, aunque ello amerita una evaluación más precisa de las variaciones del VHB.
Las 42 secuencias del estudio han tenido un alto porcentaje de similaridad con la secuencia peruana de 1993 lo cual nos indicaría que estos pacientes probablemente han estado expuestos a variantes del VHD-3 que estaban muy relacionadas. Asimismo, se observó un alto porcentaje de guanina y citocina (62%), lo cual concuerda con las referencias que indican que GC es más abundante que AT 17, con porcentajes de GC alrededor del 60% 18.
La secuencia de los 19 aminoácidos del HDAg-L es específica de cada genotipo y varía entre genotipos 19. El sitio de autoclivaje de la ribozima y el dominio de unión del ARN son muy conservados. En el extremo C-terminal se ha encontrado el motif CTQQ, el cual es el conservado en las secuencias del VHD-3, pero es muy divergente para los aac T y Q en otros genotipos 20.
La presencia de timina (T) en el nt 1014, es muy frecuente en el genotipo 3. El hallazgo de 1014C en 3 secuencias peruanas es poco frecuente, sólo hay 2 reportes similares en 1 secuencia de Brasil y 1 de Bolivia (LT604954, LT604955). En otros genotipos sí se reporta la presencia del 1014C. Este sitio es la posición de heterogeneidad que permite la formación de las 2 isoformas del HDAg mediante la edición del ARN en el codón stop con el cambio de C por la T para formar triptófano (aac195). La región 1013-1015nt corresponde al extremo final de la proteína antígeno Delta de forma corta (HDAg-S), la cual tiene función de replicación viral. La región 953-955nt corresponde al extremo final de la proteína antígeno Delta forma larga (HDAg-L) que promueve la envoltura del HDV-RNA para el ensamblaje del virión 21.
El análisis filogenético permitió determinar que las 42 secuencias reportadas de este estudio corresponden al VHD-3, lo cual es concordante con lo reportado en 1993 en un brote de hepatitis aguda en soldados asentados en cuatro puestos militares en la Amazonía del Perú 12. Este genotipo también ha sido detectado en Colombia 22, Bolivia 23, Brasil 15,19) y en Venezuela 24,25 y ha sido considerado como predominante en Sudamérica. Este reporte confirmaría la circulación del VHD-3 en el Perú, que es causante de brotes de hepatitis aguda grave 1,2.
Este estudio tiene algunas limitaciones. Las muestras utilizadas correspondieron a los años 2010 a 2012; sin embargo, brindaron información relevante del genotipo del VHD circulante en el Perú. Otra limitante fue el tiempo transcurrido desde la obtención de las muestras hasta la ejecución del estudio, esto debido a aspectos logísticos y de financiamiento. Durante este tiempo de almacenamiento se evitó el congelamiento y descongelamiento de las muestras lo cual permitió que el material genético se conserve y se pueda realizar las pruebas de n-PCR y de secuenciamiento.
El rango de sustitución nucleotídica del VHD-3 obtenido mediante el análisis Bayesiano coalescente fue similar a los obtenidos por otros autores quienes reportan rangos de 3x10-2 a 3x10-3 s/s/a 18; sin embargo, hay que considerar que estos rangos evolutivos no son homogéneos en todo el genoma, algunos consideran a 3,2x10-3 s/s/a para las región no codificante y codificante, 1,49x10-3 s/s/a para no sinónimas y 0,67x10-3 s/s/a para sinónimas 26. El tMRCA ha estimado que el VHD-3 ha estado circulando en Sudamérica desde 1930. Se tienen reportes de la presencia de la infección por VHD desde 1934 en la región amazónica 27.
En conclusión, nuestra investigación en una comunidad andina y tres amazónicas del Perú identificó el VHD-3, confirmando el predominio de este genotipo en nuestro país. Este genotipo se ha identificado en comunidades amerindias en América del Sur y probablemente se relacione con las creencias y costumbres sociales y culturales de estas poblaciones. Nuestros resultados contribuyen a mejorar el entendimiento de la coinfección VHB/VHD en el Perú.

