










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Peruana de Ginecología y Obstetricia
versión On-line ISSN 2304-5132
Rev. peru. ginecol. obstet. vol.60 no.4 Lima oct./dic. 2014
SIMPOSIO PREECLAMPSIA, VIEJO PROBLEMA AÚN NO RESUELTO: CONCEPTOS ACTUALES
Preeclampsia de inicio temprano y tardío: una antigua enfermedad, nuevas ideas
Early-and late-onset preeclampsia: an old disease, new ideas
Rommel Omar Lacunza Paredes1, José Pacheco-Romero2
1
Médico Gineco-Obstetra, Asistente del Servicio de Ginecología-Obstetricia, Hospital Nacional Daniel Alcides Carrión, Callao, Perú.2 Profesor Principal, Departamento de Obstetricia y Ginecología, Facultad de Medicina Humana, Universidad Nacional Mayor de San Marcos, Lima, Perú.
RESUMEN
Las importantes diferencias que existen entre las manifestaciones de la preeclampsia, antes (inicio temprano) y después de la semana 34 (inicio tardío), plantean la posibilidad que se trate de dos enfermedades distintas. Basada en diferencias genéticas, epidemiológicas y placentarias, esta hipótesis cobra fuerza. Sin embargo, recientes estudios nos muestran la posibilidad de una continuidad clínico temporal entre ambos fenotipos. En este contexto los fenómenos antiangiogénicos y su relación con el feto podrían ser determinantes para esclarecer la interrogante.
Palabras clave: Preeclampsia de inicio temprano, preeclampsia de inicio tardío, desbalance antiangiogénico.
ABSTRACT
Significant differences between preeclampsia manifestations before (early-onset) and after 34 weeks (late-onset) raise the possibility that these are two different diseases. This hypothesis is strengthened based on genetic, epidemiological and placental differences. However, recent studies show the possibility of temporal continuity between both clinical phenotypes. In this context antiangiogenic phenomena and their relation with the fetus may be crucial in clarifying this unanswered question.
Keywords: Early-onset preeclampsia, late-onset preeclampsia, antiangiogenic imbalance.
INTRODUCCIÓN
Desde la definición de preeclampsia, en 2001, fue evidente el afán de la simplificación del diagnóstico con el objetivo de lograr un mejor y oportuno manejo. Sin embargo, la búsqueda de una definición que refleje claramente la naturaleza multisistémica de lo que entendemos hoy por preeclampsia continúa.
La pregunta inicial sería qué es la preeclampsia, ¿únicamente la elevación de la presión arterial en la gestante asociada a proteinuria o estos son es sí mismos solo signos inespecíficos del verdadero trastorno (disfunción endotelial)? Se debe tomar en cuenta que el aumento de la presión arterial y la proteinuria pueden ser encontradas en otras entidades, como es el caso de la proteinuria gestacional, hipertensión gestacional transitoria, síndrome en espejo, entre otros(1).
La publicación de la nueva definición de preeclampsia por parte del American Congress of Obstetricians and Gynecologists (ACOG) muestra el esfuerzo por integrar estos conceptos y revalorizar la afectación multisistémica en la gestante, dejando de lado la necesidad de evidenciar proteinuria y aceptando signos equivalentes de daño de órganos como son la plaquetopenia, alteración hepática, afectación renal, edema pulmonar y disturbios visuales o neurológicos(2). Sin embargo, esta definición aún nos obliga a encontrar hipertensión arterial manifiesta, la cual es ya un signo tardío de la enfermedad.
Recientemente, la revalorización de antiguas investigaciones y los nuevos descubrimientos sugieren clasificar la preeclampsia en dos tipos, fenotipos o clases de manifestación clínica, como preeclampsia de inicio precoz (PIP) y de inicio tardío (PIT), con un punto de corte a las 34 semanas(3). Esto obliga a revisar las bases para esta diferenciación. El objetivo del presente artículo es mostrar al lector la evidencia publicada hasta el momento, revisando algunos conceptos comunes en la fisiopatología. No se pretende profundizar los aspectos moleculares, que son mucho mejor explicados en otras revisiones.
LAS DIFERENCIAS EPIDEMIOLÓGICAS
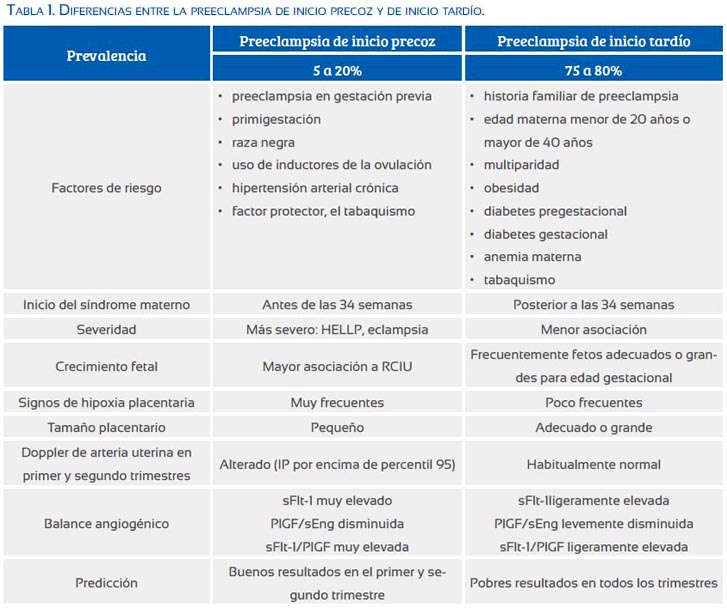
Como primer punto relevante se debe mencionar que la PIT constituye entre 75 y 80% de los casos de preeclampsia, mientras la PIP entre 5 y 20%, según sea la serie publicada(4,5). Recientemente, Lisonkova (6) comunica una prevalencia de 0,38 y 2,72 casos por cada 100 partos de PIP y PIT, respectivamente.
Si bien existen factores de riesgo comunes a ambas entidades, la mayoría de estudios coincide en que existe una mayor asociación de la PIT con historia familiar de preeclampsia, edad materna menor de 20 años o mayor de 40 años, multiparidad, obesidad, mayor ganancia de peso durante la gestación, diabetes gestacional, anemia materna y tabaquismo. Para el caso de la PIP, existe mayor asociación con preeclampsia en gestación previa, primigestación, raza negra, uso de inductores de la ovulación y, como factor protector, el tabaquismo(6-12). En el caso particular de la diabetes pregestacional, se encuentra incremento del riesgo para ambos tipos, pero en mayor grado para PIT; todo lo contrario sucede con la hipertensión crónica, que aumenta considerablemente el riesgo para PIP (6) (tabla 1).
LAS DIFERENCIAS CLÍNICAS
Los cuadros clínicos más severos se observan en los casos de PIP, mostrando mayor asociación con eclampsia, síndrome de HELLP, falla multisistémica, RCIU y fetos pequeños para edad gestacional, con el consiguiente aumento de la morbimortalidad materno fetal(3,11,13). A diferencia, los casos de inicio tardío suelen ser más benignos, sin llegar a presentar cuadros severos en la mayoría de ocasiones(14) y los recién nacidos suelen tener peso adecuado o son grandes para la edad gestacional(3,11).
También se ha descrito diferencias hemodinámicas, encontrando en la PIP aumento de la resistencia periférica, con respuesta baja del gasto cardiaco, y en la PIT, resistencia periférica disminuida con gasto cardiaco aumentado, en lo que algunos investigadores denominan la fase latente de la preeclampsia(15,16).
LA INVASIÓN TROFOBLÁSTICA DEFICIENTE, LA HIPOPERFUSIÓN PLACENTARIA Y LA PREDICCIÓN
Es aceptada actualmente en la fisiopatología de la preeclampsia la deficiente invasión trofoblástica de los vasos espirales(17), pero también es conocido que no es posible demostrarla en todos los casos que desarrollan esta enfermedad.
Se conoce que la invasión deficiente produce fenómenos en los vasos sanguíneos y en la circulación placentaria, que ocasionan hipoperfusión (18), mostrando en el estudio anatomopatológico menor volumen placentario, disminución de la superficie de las vellosidades coriales, aumento del número de infartos, arteriopatía decidual e hipermaduración de las vellosidades coriales(13,19-21).
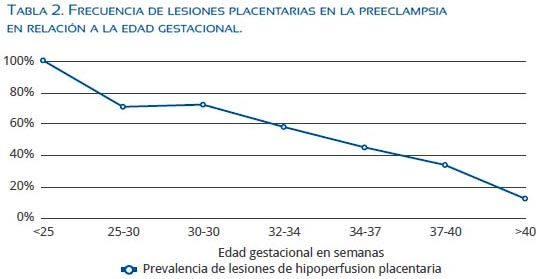
Las gestaciones afectadas por preeclampsia antes de las 34 semanas presentan con gran frecuencia signos de hipoperfusión placentaria, encontrándose a una menor edad gestacional de presentación de la preeclampsia mayor frecuencia de lesiones(13) (tabla 2), porcentaje que se incrementa si se asocia a RCIU(21). En contraste, la frecuencia de lesiones placentarias asociadas a hipoxia en la PIT es mucho más baja, con tamaños placentarios que suelen ser normales o mayores a los normales, como se ha señalado en varias series(3,22). Un reciente estudio evaluó la fracción de perfusión placentaria, encontrando en la PIP una disminución significativa en comparación a la PIT, donde la fracción de perfusión fue mayor o igual a la encontrada en gestaciones normales(23). Todo esto apoya la importancia de la hipoperfusión placentaria en la génesis de la PIP; en cambio, en la PIT parece no cumplir un rol tan trascendental(24).
La hipoperfusión placentaria y el estrés oxidativo en el espacio intervelloso por la deficiente invasión trofoblástica en la preeclampsia producen la expresión de múltiples moléculas que han sido utilizadas en los intentos de predicción, aisladas o en combinación con el estudio Doppler de las arterias uterinas, la presión arterial media y con factores epidemiológicos maternos(25-31).
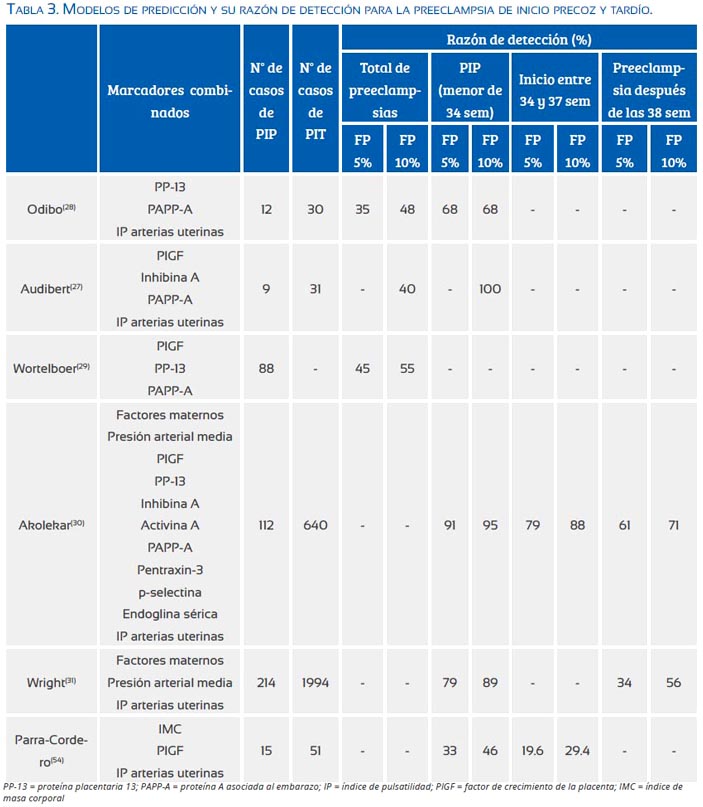
Una de las moléculas predictoras más prometedoras es la PP-13 (placental protein), la cual es únicamente producida en la placenta, con funciones tanto en el desarrollo como en el remodelado vascular de las arterias espirales en la invasión trofoblástica; su disminución en el primer trimestre ha sido relacionada a la aparición de la PIP, en el trabajo de Romero(32), y con la necesidad de término antes de las 34 semanas en la gestación asociada a preeclampsia, en el estudio de Nicolaides(33).
El aumento de la resistencia de las arterias uterinas en el primer(34) y segundo trimestre(35) ha sido vinculado a la invasión trofoblástica deficiente, siendo señalado como un factor predictor primordialmente para la PIP.
Lo anteriormente expuesto podría explicar por qué los modelos actuales que han evaluado múltiples moléculas en el primer trimestre, como PP-13, PAPP-A (Pregnancy associated protein A), PlGF (placental growth factor), inhibina-A, activina-A, endoglina sérica (sEng), pentanxin-3 y ADAM12 (A disintegrin and metalloprotease 12), muestran pobre capacidad predictiva para la PIT(27-31). Sin embargo, los resultados aún son difíciles de interpretar debido a la multiplicidad de definiciones y puntos de corte utilizados (tabla 3). Podemos suponer que los intentos de predecir PIT basados en marcadores de la deficiente invasión trofoblástica y la hipoperfusión placentaria han fracasado al no ser factores fisiopatológicos gravitantes(3,13).
Finalmente, la placenta (con una deficiente invasión trofoblástica) ante estados de hipoperfusión prolongados y estrés oxidativo en el caso de la PIP generaría moléculas que llevan a la disfunción endotelial materna(17,18,36). Pero, en la PIT la evidencia de estados de hipoperfusión placentaria no es frecuente y, como se mencionó, las placentas no muestran estos cambios al estudio anatomo-patológico(13,19-21), quedando la interrogante sobre qué es lo que desencadena la disfunción endotelial en la PIT.
LAS DIFERENCIAS MOLECULARES: LA DISFUNCIÓN ENDOTELIAL Y EL SÍNDROME ANTIANGIOGÉNICO MATERNO
En los últimos años, la búsqueda incesante de la sustancia definitiva que explique los fenómenos que acontecen en la gestante con preeclampsia ha sido infructuosa y esto podría deberse a la existencia de distintas vías metabólicas que explican los mismos signos fundamentales, tanto en la preeclampsia de inicio tardío como de inicio precoz.
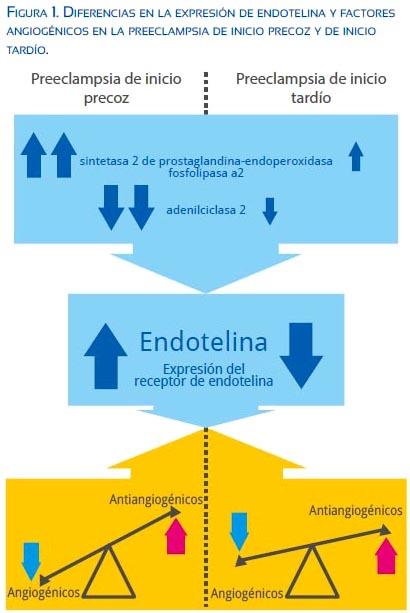
La expresión genética placentaria de la endotelina y sus efectos sobre la disfunción endotelial en la preeclampsia muestran importantes diferencias entre la PIP y la PIT. Sitras(37) encuentra que la sintetasa 2 de prostaglandina-endoperoxidasa con la fosfolipasa A2 están sobreexpresadas y en cambio la adenilciclasa 2 está infraexpresada en la PIP en comparación con la PIT. Dieber-Rotheneder(38) comunica que la endotelina sérica materna y su receptor (importantes mediadores de la hipertensión arterial) están elevados en la PIP y más aún en los casos asociados a RCIU; sin embargo, los niveles de endotelina en la PIT han sido encontrados claramente disminuidos (figura 1). Esta evidencia en conjunto podría sugerir entonces que se trata de dos entidades distintas, como proponen los autores(39).
En lo referente a los factores angiogénicos, la expresión de TGF beta (transforming growth factor) y VEGF 1-2 (vascular endothelial growth factor), que están involucrados en regular el adecuado desarrollo vascular placentario(40) , ha mostrado ser bastante similar en ambos tipos de preeclampsia(24,37).
En los últimos años se ha demostrado la existencia de un desbalance angiogénico en las gestantes con preeclampsia, con aumento de sustancias antiangiogénicas (sFlt-1, sEng)(1,3,40-42). El sFlt-1 (soluble Fms-like tyrosine kinase) constituye el receptor soluble del VEGF-1; impide su unión con el receptor de membrana bloqueando su acción angiogénica. De igual forma, la endoglina sérica (sEng) impide la unión del TGF-beta a su receptor celular(1,40). Estas producirían la disfunción endotelial y finalmente el síndrome materno, tal y como lo han mostrado los estudios que al administrar sFlt-1 en ratas preñadas producen elevación de la presión arterial y endoteliosis glomerular renal(43-45). También se ha encontrado que la elevación de los niveles séricos maternos de sFlt-1 precede el inicio del cuadro clínico materno en aproximadamente 1 a 2 meses(3,40).
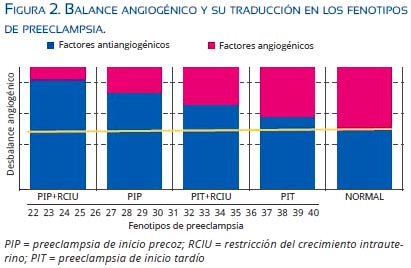
La producción de sFlt-1 es estimulada por la hipoxia placentaria a través de la expresión de HIF-1 (hypoxia inducible factor)(46) y ello explica por qué los niveles son más altos en la PIP(3,21) (figura 1). Wikstrom(47) encuentra elevación en 43 veces de los niveles de sFlt-1 en la PIP y de solo 3 veces en la PIT, con respecto a controles sanos. Recientemente, Tobinaga(48) halló diferencias significativas entre las 28 y 36 semanas de los niveles de sFlt-1 y sEng en la PIT versus la PIP. El grado de desequilibrio angiogénico podría determinar las manifestaciones clínicas del cuadro materno fetal, indudablemente influido por la carga genética y los factores ambientales(49) (figura 2), lo que sugeriría que, si bien fueran dos enfermedades distintas, compartirían una vía metabólica final común, produciendo un cuadro clínico similar (compromiso materno multiorgánico producido por disfunción endotelial).
Por otro lado, la valoración de los factores angiogénicos en el segundo(50) y tercer trimestre se presentan como prometedoras para la predicción de la PIT. Recientemente, Chaiworapomgsa(51) informa que la reducción de la relación sérica de PlGF/sFlt-1 y PlGF/sEng entre las 30 y 34 semanas de gestación demuestra ser buena predictora de la PIT. De igual forma, Lai relaciona la disminución del PIGF y la elevación del sFlt-1 entre las 30 y 33 semanas con el inicio en 4 a 8 semanas de la preeclampsia(52). Oliveira(53), al evaluar distintos algoritmos para la predicción de la PIT en el primer trimestre, encuentra que el modelo propuesto por Parra-Cordero(54) tiene el mejor rendimiento y esto se podría explicar a que incluye factores maternos (IMC, IP de la arteria uterina ) y biomarcadores como el PlGF (placental growth factor).
Aún queda por dilucidar cuál es el estímulo que produce este estado antiangiogénico en la PIT, considerando que la invasión trofoblástica ha sido adecuada y la hipoxia placentaria no es un factor determinante; qué otros factores podrían producir de la disfunción endotelial y el síndrome materno en la PIT es aún la gran pregunta.
LA IMPORTANCIA DE LA INFLAMACIÓN CRÓNICA EN LA PIT
En la patogénesis de la preeclampsia existe una compleja alteración de la función inmunológica, con un estado de hiperrespuesta inmune materna(17,55). En la preeclampsia hay aumento de las sustancias inflamatorias; lo que no está determinado con claridad es si ello es consecuencia o causa de la disfunción endotelial. De igual forma, la inflamación coincide con la alteración inmunológica compleja que ocurre en la preeclampsia desde la implantación. Queda aún por dilucidar su relación con el estado antiangiogénico materno(1,3,5,40).
Un factor proinflamatorio propuesto son las micropartículas de sincitiotrofoblasto circulantes en sangre materna (56). Goswami(57) encontró un mayor número de micropartículas en las gestantes con PIP; en comparación, los valores en la PIT no difirieron mayormente de las gestaciones normales. Esto indicaría en algún grado la contribución de las micropartículas de sincitiotrofoblasto en la génesis de la PIP y no en la PIT(3).
La fuerte asociación epidemiológica que existe entre la obesidad(54), obesidad mórbida(10) y la PIT es tentativamente explicada por existir un estado materno de inflamación crónica leve producido por las múltiples moléculas inflamatorias liberadas por los adipocitos(5), lo cual en determinadas circunstancias, aún no claramente establecidas, podría contribuir o desencadenar la disfunción endotelial y el síndrome materno en la PIT.
La hipótesis que algunos proponen es que, ante un estado de inflamación crónica leve, una mínima elevación de las sustancias antiangiogénicas desencadena la disfunción endotelial y el síndrome materno en el caso de la PIT(5).
VALOR DE LA INSUFICIENCIA PLACENTARIA RELATIVA: EL ORIGEN FETAL DE LA PIT
La hipoperfusión placentaria se constituye en el fenómeno fisiopatológico esencial en la génesis de la PIP, ocasionando la liberación de sustancias antiangiogénicas por la placenta, produciendo disfunción endotelial y el síndrome materno(17). Esta hipoperfusión placentaria también se presenta en los casos de enfermedad trofoblástica (placenta con degeneración hidrópica)(1,58), comportándose como el ejemplo más extremo de insuficiencia placentaria, con gran producción de sustancias antiangiogénicas debido a la hipoxia, generando en algunos casos un cuadro clínico similar a la preeclampsia, con hipertensión y proteinuria antes de las 20 semanas de gestación(1).
El momento en el cual se inicia la hipoperfusión placentaria desencadenaría el desequilibrio angiogénico. A menor edad gestacional de inicio, esto sería más severo, generando el cuadro clínico típico de la PIP(49).
El concepto de insuficiencia placentaria relativa hace alusión a la incapacidad de la placenta para suplir los requerimientos fetales. Podríamos imaginar un escenario en el cual un feto requiere mayor aporte del que está recibiendo (como en la macrosomía fetal o en fetos de madres diabéticas) y entrar en insuficiencia placentaria relativa (como algunos investigadores proponen)(1,58). Entonces, es necesario, de alguna forma, que el feto manifieste este estado de carencia e intente mejorarlo a través de la producción de moléculas que mejoren la perfusión placentaria, siendo estas las que desencadenan el desequilibrio angiogénico(1).
Se propone así que la adenosina producida por el feto es la molécula que mejoraría la perfusión placentaria, aumentando el gasto cardiaco y elevando la presión arterial materna en los casos de preeclampsia por encima de las 34 semanas(59); de esta forma intentaría compensar la insuficiencia placentaria relativa.
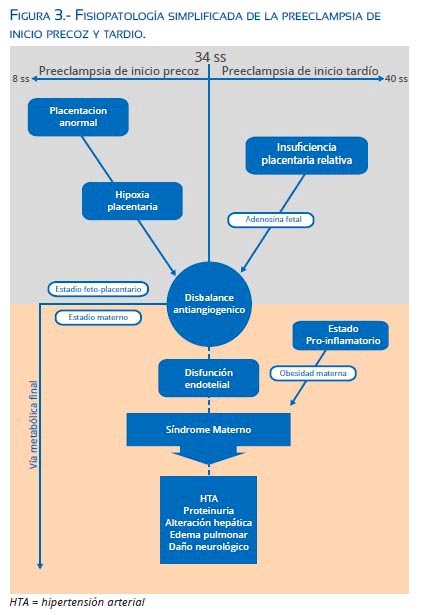
El aumento de la adenosina fetal incrementa el sFlt1 por efecto de la sobreexpresión de HIF-1 y disminuye el PlGF, generando desequilibrio angiogénico, lo cual ocurre existiendo o no hipoxia placentaria(1,58,59). Espinoza sintetiza esta hipótesis y propone el síndrome de respuesta antiangiogénica materno fetal, que explicaría el inicio del síndrome materno en los casos de PIT(1) (figura 3).
UNA SOLA ENFERMEDAD, UNA CONTINUIDAD EVOLUTIVA
Recientemente, el grupo del profesor Nicolaides publica un modelo de supervivencia que intenta explicar la pobre capacidad predictiva de los marcadores en la PIT(31). En su modelo matemático se considera que teóricamente las gestaciones de riesgo alto desarrollarían preeclampsia severa antes de las 34 semanas, con una probabilidad entre 0,6 y 60%. Para los casos de población de riesgo bajo, la probabilidad antes de las 34 semanas sería de 0,01 a 1%. Considerando teóricamente la prolongación indeterminada de la gestación, en los casos de riesgo bajo la mayoría podría desarrollar preeclampsia por encima de las 40 semanas (una media de 55 semanas con una desviación estándar de 7 semanas), según el modelo matemático; proponiendo entonces que la PIP y PIT son en realidad estadios temporales de la misma enfermedad(60).
El escenario sería un extremo de la enfermedad poco frecuente (20%), severo y con rápida progresión para el caso de la PIP, con gran desequilibrio angiogénico, manifestado principalmente antes de las 34 semanas, versus una evolución más lenta y benigna en 80% de los casos para la PIT, con un menor grado de desequilibrio angiogénico manifestado al término de las gestación (lo cual correspondería al extremo de curva de frecuencia propuesta por Nicolaides). Esta diferencia en su evolución podría estar determinada por la influencia de otros factores genéticos maternos o placentarios. Sin embargo, esta afirmación aún requiere posteriores estudios que demuestren esta continuidad fisiopatológica.
Es indudable que tanto la PIP como la PIT comparten una base genética común. Sin embargo, la diferencia final fenotípica estaría dada por la interacción de genes particulares. Triche(61), en su aproximación bioinformática de la genética de la preeclampsia, pone en evidencia el escaso conocimiento de los genes maternos y fetales relacionados a la PIT.
CONCLUSIONES
A pesar de los grandes avances en el entendimiento de la fisiopatología del síndrome de preeclampsia, queda aún mucho por dilucidar. La discusión sobre si la PIP y la PIT son dos enfermedades o una evolución de la misma está aún abierta, con muchas interrogantes por responder. No debemos olvidar que en nuestro medio la enfermedad hipertensiva del embarazo es una causa muy importante de muerte materna y ningún esfuerzo es innecesario tanto para su comprensión como para su manejo y prevención. El mejor conocimiento de la PIT nos ayudará a mejorar nuestra atención en la práctica diaria.
AGRADECIMIENTOS
A mi esposa (RL), Naida por su inmensa paciencia e inagotable apoyo en todos los aspectos de mi vida.
Al Dr. Jorge Avalos Gómez por su valiosa colaboración con los gráficos.
REFERENCIAS BIBLIOGRÁFICAS
1. Espinoza J, Uckele JE, Starr RA, Seubert DE, Espinoza AF, Berry SM. Angiogenic imbalances: the obstetric perspective. Am J Obstet Gynecol 2010; 203:17.e1-8. doi: 10.1016/j.ajog.2009.10.891.
2. American College of Obstetricians and Gynecologists. Task force on Hypertension in pregnancy. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists Task Force on Hypertension in Pregnancy. Obstet Gynecol. 2013 Nov;122(5):1122-31. doi: 10.1097/01.AOG.0000437382.03963.88.
3. Raymond D, Peterson E. A critical review of early-onset and late-onset preeclampsia. Obstet Gynecol Surv. 2011 aug;66(8):497-506. doi: 10.1097/OGX.0b013e3182331028.
4. Huppertz B. Placental origins of preeclampsia: challenging the current hypothesis. Hypertension. 2008 Apr;51(4):970-5. doi: 10.1161/HYPERTENSIONAHA.107.107607.
5. Ferrazzi E, Stampalija T, Aupont JE. The evidence for late onset preeclampsia as a maternogenic disease of pregnancy. Fetal Matern Med Rev. 2013 Feb;24(1):18-31. DOI: http://dx.doi.org/10.1017/S0965539513000028.
6. Lisonkova S, Joseph KS. Incidence of preeclampsia: risk factors and outcomes associated with early- versus late-onset disease. Am J Obstet Gynecol. 2013 Dec;209(6):544.e1-12. doi: 10.1016/j.ajog.2013.08.019.
7. Ornaghi S, Tyurmorezova A, Algeri P, Giardini V, Ceruti P, Vertemati E, Vergani P. Influencing factors for late-onset preeclampsia. J Matern Fetal Neonatal Med. 2013 Sep;26(13):1299-302. doi: 10.3109/14767058.2013.783807.
8. Odegard RA, Vatten LJ, Nilsen ST, Salvesen KA, Austgulen R. Risk factors and clinical manifestations of pre-eclampsia. BJOG. 2000 Nov;107(11):1410-6.
9. Poon LC, Kametas NA, Chelemen T, Leal A, Nicolaides KH. Maternal risk factors for hypertensive disorders in pregnancy: a multivariate approach. J Hum Hypertens. 2010 Feb;24(2):104-10. doi: 10.1038/jhh.2009.45.
10. Aksornphusitaphong A, Phupong V. Risk factors of early and late onset pre-eclampsia. J Obstet Gynaecol Res. 2013 MAr;39(3):627-31. doi: 10.1111/j.14470756.2012.02010.x.
11. Phillips J, Janowiak M, Badger G, Bernstein I. Evidence for distinct preterm and term phenotypes of preeclampsia. J Matern Fetal Neonatal Med. 2010 Jul;23(7):622-6. doi: 10.3109/14767050903258746.
12. Sohlberg S, Stephansson O, Cnattingius S, Wikstrom AK. Maternal body mass index, height, and risks of preeclampsia. Am J Hypertens 2012 Jan;25(1):120-5. doi: 10.1038/ajh.2011.175.
13. Ogge G, Chaiworapongsa T, Romero R, Hussein Y, Kusanovic JP, Yeo L, Kim CJ, Hassan S. Placental lesions associated with maternal underperfusion are more frequent in early-onset than in late-onset preeclampsia. J Perinat Med. 2011 Nov;39(6):641-52. doi: 10.1515/ JPM.2011.098.
14. Kenneth L, Hall DR, Gebhardt S, Grove D. Late onset preeclampsia is not an innocuous condition. Hypertens Pregnancy. 2010;29(3):262-70. doi: 10.3109/10641950902777697.
15. Valensise H, Novelli GP, Vasapollo B. Pre-eclampsia: one name, two conditions - the case for early and late disease being different. Fetal Matern Med Rev. 2013;24(1):32-37. DOI: 10.1017/S0965539513000016.
16. Valensise H, Vasapollo B, Gagliardi G, Novelli GP. Early and late preeclampsia: two different maternal hemodynamic states in the latent phase of the disease. Hypertension. 2008 Nov;52(5):873-80. doi: 10.1161/HYPERTENSIONAHA.108.117358.
17. Palei A, Spradley F, Warrington J, George E, Granger J. Pathophysiology of hypertension in pre-eclampsia: a lesson in integrative physiology. Acta Physiol. 2013 Jul;208(3):224-33. DOI: 10.1111/apha.12106.
18. Redman C, Sargent I. Placental stress and pre-eclampsia: a revised view. Placenta. 2009 Mar;23 Suppl A:S38-42. doi: 10.1016/j.placenta.2008.11.021.
19. Moldenhauer JS, Stanek J,Warshak C, Khoury J, Sibai B. The frequency and severity of placental findings in women with preeclampsia are gestational age dependent. Am J Obstet Gynecol. 2003 Oct;189(4):1173-7.
20. van der Merwe JL, Hall DR, Wright C, Schubert P, Grové D. Are early and late preeclampsia distinct subclasses of the diseasewhat does the placenta reveal? Hypertens Pregnancy. 2010; 29:457-67. doi: 10.3109/10641950903572282.
21. Kovo M, Schreiber L, Ben-Haroush A, Gold E, Golan A, Bar J. The placental component in early-onset and late-onset preeclampsia in relation to fetal growth restriction. Prenat Diagn. 2012 Jul;32(7):632-7. doi: 10.1002/pd.3872.
22. Nelson DB, Ziadie MS, McIntire DD, Rogers BB, Leveno KJ. Placental pathology suggesting that preeclampsia is more than one disease. Am J Obstet Gynecol. 2014 Jan;210(1):66.e1-7. doi: 10.1016/j.ajog.2013.09.010.
23. Sohlberg S, Mulic-Lutvica A, Lindgren P, Ortiz-Nieto F, Wikström AK, Wikström J. Placental perfusion in normal pregnancy and early and late preeclampsia: A magnetic resonance imaging study. Placenta. 2014 Mar;35(3):202-6. doi: 10.1016/j.placenta.2014.01.008.
24. Soto E, Romero R, Kusanovic JP, Ogge G, Hussein Y, Yeo L, Hassan SS, Kim CJ, Chaiworapongsa T. Late-onset preeclampsia is associated with an imbalance of angiogenic and anti-angiogenic factors in patients with and without placental lesions consistent with maternal underperfusion. J Matern Fetal Neonatal Med. 2012 May;25(5):498-507. doi: 10.3109/14767058.2011.591461.
25. Anderson UD, Olsson MG, Kristensen KH, Akerström B, Hansson SR. Review: Biochemical markers to predict preeclampsia. Placenta. 2012 Feb;33 Suppl:S42-7. doi: 10.1016/j.placenta.2011.11.021.
26. Monte S. Biochemical markers for prediction of preclampsia: review of the literature. J Prenat Med. 2011 Jul;5(3):69-77.
27. Audibert F, Boucoiran I, An N, Aleksandrov N, Delvin E, Bujold E, Rey E. Screening for preeclampsia using first-trimester serum markers and uterine artery Doppler in nulliparous women. Am J Obstet Gynecol. 2010 Oct;203(4):383 e1-8. doi: 10.1016/j.ajog.2010.06.014.
28. Odibo AO, Zhong Y, Goetzinger KR, Odibo L, Bick JL, Bower CR, et al. First trimester placental protein 13, PAPP-A, uterine artery Doppler and maternal characteristics in the prediction of pre-eclampsia. Placenta. 2011 Aug;32(8):598-602. doi: 10.1016/j.placenta.2011.05.006.
29. Wortelboer EJ, Koster MP, Cuckle HS, Stoutenbeek PH, Schielen PC, Visser GH. First-trimester placental protein 13 and placental growth factor: markers for identification of women destined to develop early-onset pre-eclampsia. BJOG. 2010 Oct;117(11):1384-9. doi: 10.1111/j.1471-0528.2010.02690.x.
30. Akolekar R, Syngelaki A, Sarquis R, Zvanca M, Nicolaides KH. Prediction of early, intermediate and late pre-eclampsia from maternal factors, biophysical and biochemical markers at 11-13 weeks. Prenat Diagn. 2011 Jan;31(1):66-74. doi: 10.1002/pd.2660.
31. Wright D, Akolekar R, Syngelaki A, Poon LC, Nicolaides KH. A competing risks model in early screening for preeclampsia. Fetal Diagn Ther. 2012;32(3):171-8. doi: 10.1159/000338470.
32. Romero R, Kusanovic JP, Than NG, Erez O, Gotsch F, Espinoza J, et al. First trimester maternal serum PP13 in the risk assessment for preeclampsia. Am J Obstet Gynecol. 2008 Aug;199(2):122 e1-11. doi: 10.1016/j.ajog.2008.01.013.
33. Nicolaides KH, Bindra R, Turan OM, Chefetz I, Sammar M, Meiri H, et al. A novel approach to first-trimester screening for early pre-eclampsia combining serum PP-13 and Doppler ultrasound. Ultrasound Obstet Gynecol. 2006 Jan;27(1):13-7.
34. Papageorghiou AT, Campbell S. First trimester screening for preeclampsia. Curr Opin Obstet Gynecol. 2006 Dec;18(6):594-600.
35. Papageorghiou AT, Yu CK, Cicero S, Bower S, Nicolaides KH. Second-trimester uterine artery Doppler screening in unselected populations: a review. J Matern Fetal Neonatal Med. 2002 Aug;12(2):78-88.
36. Poston L. Endothelial dysfunction in pre-eclampsia. Pharmacol Rep. 2006;58:69-74.
37. Sitras V, Paulssen RH, Gronaas H, Leirvik J, Hanssen TA, Vartun A, Acharya G. Differential placental gene expression in severe preeclampsia. Placenta. 2009 May;30(5):424-33. doi: 10.1016/j.placenta.2009.01.012.
38. Dieber-Rotheneder M, Beganovic S, Desoye G, Lang U, Cervar-Zivkovic M. Complex expression changes of the placental endothelin system in early and late onset preeclampsia, fetal growth restriction and gestational diabetes. Life Sci. 2012 Oct 15;91(13-14):710-5. doi: 10.1016/j.lfs.2012.04.040.
39. Roberts JM, Catov JM. Preeclampsia more than 1 disease: or is it? Hypertension. 2008 Apr;51(4):989-90. doi: 10.1161/HYPERTENSIONAHA.107.100248.
40. Maynard S, Epstein FH, Karumanchi SA. Preeclampsia and angiogenic imbalance. Annu Rev Med 2008;59:61-78.
41. Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, Sibai BM, Epstein FH, Romero R, Thadhani R, Karumanchi SA. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006 Sep 7;355(10):992-1005.
42. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003 Mar;111(5):649-58.
43. Kim YN, Lee DS, Jeong DH, sung MS, Kim KT. The relationship of the level of circulating antiangiogenic factors to the clinical manifestations of preeclampsia. Prenat Diagn. 2009 May;29(5):464-70. doi: 10.1002/ pd.2203.
44. Oudejans CB, van Dijk M, Oosterkamp M, Lachmeijer A, Blankenstein MA. Genetics of preeclampsia: paradigm shifts. Hum Genet. 2007 Jan;120(5):607-12.
45. Gilbert JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension. 2007 Dec;50(6):1142-7.
46. Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A, Zamudio S, Caniggia I. Increased expression of sFlt1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol. 2006 Oct;291(4):1085-93.
47. Wikstrom AK, Larsson A, Eriksson UJ, Nash P, Nordén-Lindeberg S, Olovsson M. Placental growth factor and soluble fms-like tyrosine kinase-1 in early-onset and late-onset preeclampsia. Obstet Gynecol. 2007 Jun;109(6):1368-74.
48. Tobinaga CM, Torloni MR, Gueuvoghlanian-Silva BY, Pendeloski KPT, Akita PA, Sass N, Daher S. Angiogenic factors and uterine doppler velocimetry in early- and late-onset preeclampsia. Acta Obstet Gynecol Scand. 2014 May;93(5):469-76. doi: 10.1111/aogs.12366.
49. Espinoza J. Angiogenic imbalances in the pathogenesis of pregnancy complications. Fetal Matern Med Rev. 2014 Feb;25(1):42-58. doi.org/10.1017/S0965539514000096.
50. Erez O, Romero R, Espinoza J, Fu W, Todem D, Kusanovic JP, Gotsch F, Edwin S, Nien JK, Chaiworapongsa T, Mittal P, Mazaki-Tovi S, Than NG, Gomez R, Hassan SS. The change in concentrations of angiogenic and anti-angiogenic factors in maternal plasma between the first and second trimesters in risk assessment for the subsequent development of preeclampsia and smallfor- gestational age. J Matern Fetal Neonatal Med. 2008 May;21(5):279-87. doi: 10.1080/14767050802034545.
51. Chaiworapongsa T, Romero R, Korzeniewski SJ, Kusanovic JP, Soto E, Lam J, Dong Z, Than NG, Yeo L, et al. Maternal plasma concentrations of angiogenic/ antiangiogenic factors in the third trimester of pregnancy to identify the patient at risk for stillbirth at or near term and severe late preeclampsia. Am J Obstet Gynecol. 2013 Apr;208(4):287.e1-15. doi: 10.1016/j. ajog.2013.01.016.
52. Lai J, Garcia-Tizon Larroca S, Peeva G, Poon LC, Wright D, Nicolaides KH. Competing risks model in screening for preeclampsia by serum placental growth factor and soluble fms-Like tyrosine kinase-1 at 30-33 weeks gestation. Fetal Diagn Ther. 2014;35(4):240-8. doi: 10.1159/000359968.
53. Oliveira N, Magder LS, Blitzer MG, Baschat AA. First-trimester prediction of pre-eclampsia: external validity of algorithms in a prospectively enrolled cohort. Ultrasound Obstet Gynecol. 2014 Sep;44(3):279-85. doi: 10.1002/uog.13435.
54. Parra-Cordero M, Rodrigo R, Barja P, Bosco C, Rencoret G, Sepulveda-Martinez A, Quezada S. Prediction of early and late pre-eclampsia from maternal characteristics, uterine artery Doppler and markers of vasculogenesis during first trimester of pregnancy. Ultrasound Obstet Gynecol. 2013 May;41(5):538-44. doi: 10.1002/uog.12264.
55. Borzychowski AM, Sargent IL, Redman CW. Inflammation and pre-eclampsia. Semin Fetal Neonatal Med. 2006 Oct;11(5):309-16.
56. Lee SM, Romero R, Lee YJ, Park IS, Park CW, Yoon BH. Systemic inflammatory stimulation by microparticles derived from hypoxic trophoblast as a model for inflammatory response in preeclampsia. Am J Obstet Gynecol. 2012 Oct;207(4):337e1-8. doi: 10.1016/j. ajog.2012.06.047.
57. Goswami D, Tannetta DS, Magee LA, Fuchisawa A, Redman CW, Sargent IL, von Dadelszen P. Excess syncytiotrophoblast microparticle shedding is a feature of early onset preeclampsia, but not normotensive intrauterine growth restriction. Placenta. 2006 Jan;27(1):56-61.
58. Espinoza J. Uteroplacental ischemia in early- and late-onset pre-eclampsia: a role for the fetus? Ultrasound Obstet Gynecol. 2012 Oct;40(4):373-82. doi: 10.1002/uog.12280.
59. Espinoza J, Espinoza AF, Power GG. High fetal plasma adenosine concentration: a role for the fetus in preeclampsia? Am J Obstet Gynecol. 2011 Nov;205(5):485. e24-7. doi: 10.1016/j.ajog.2011.06.034.
60. Poon LC, Nicolaides KH. First-trimester maternal factors and biomarker screening for preeclampsia. Prenat Diagn. 2014 Jul;34(7):618-27. doi: 10.1002/pd.4397.
61. Triche E, Uzun A, DeWan AT, Kurihara I, Liu J, Occhiogrosso R, Shen B, Parker J, Padbury JF. Bioinformatic approach to the genetics of preeclampsia. Obstet Gynecol. 2014 Jun;123(6):1155-61. doi: 10.1097/AOG.0000000000000293.
Fuente de financiamiento: Autofinanciado
Conflictos de interés: Ninguno.
Correspondencia:
Dr. Rommel Omar Lacunza Paredes.
Dirección: Jr. Genaro Delgado Mz. K Lt. 9A, San Martin de Porres, Lima.
Celular: 985436784
drrlacunza@hotmail.com

