











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO  uBio
uBio 
 Permalink
Permalink
Introducción
En las últimas décadas el cóndor andino (Vultur gryphus Linnaeus, 1758) ha sufrido un alarmante descenso poblacional, probablemente debido a causas antropogénicas como la caza y la expansión de la frontera agrícola, siendo considerada una especie amenazada, actualmente considerada en el apéndice I de la Convención sobre el Comercio Internacional de Especies Amenazadas de Fauna y Flora Silvestres (CITES 2020), en el apéndice II de la Convención sobre la conservación de las especies migratorias de animales silvestres (CMS 2020); en la lista roja de la Unión Internacional para la Conservación de la Naturaleza (IUCN) es considerado como vulnerable (VU) (BirdLife International 2020). En el Perú tiene la categoría de especie "En Peligro" (EN) por D.S. N° 004-2014-MINAGRI y la Ley N° 30203 promulgada el 10 de junio de 2014 determina de interés nacional y necesidad pública la protección y conservación del cóndor andino.
Se conoce que la reducción del número de individuos de una población silvestre puede conllevar a problemas genéticos que la ponen en peligro. Uno de los problemas más comunes es la pérdida de la diversidad genética que podrían conllevar a difundirse en la población alteraciones genéticas negativas por la alta endogamia, o que se produzca una disminución e incluso pérdida de alelos, lo que causaría una reducción en la viabilidad de los individuos en futuras generaciones (Hendrickson et al. 2003, Adams & Villablanca 2007), y la probabilidad de la reducción de las combinaciones posibles de genes capaces de conferir adaptación ante las variaciones ambientales (Piñero et al. 2008).
A pesar que en las últimas décadas los estudios genéticos han contribuido a las medidas de conservación de varias especies (Barrowclough et al. 1999, Asai et al. 2006, Agudo 2011, Arguello & García 2014), son pocos los estudios genéticos realizados en cóndores andinos. En el Ecuador se realizó la determinación de parentesco de cóndores en cautiverio utilizando análisis de ADN con la finalidad de reintroducirlos en su territorio y evitar la endogamia y la deriva genética propia de poblaciones pequeñas. Así, el análisis de genotipo y la caracterización de 19 individuos en base a seis microsatélites con variaciones de 93 a 220 pb reveló un nivel de heterocigosidad aceptable para 5 microsatélites. La prueba fue suficiente para diferenciar entre individuos, pero no describe con alta precisión la relación de parentesco entre los individuos (Navarrete 2012).
Con el propósito de evaluar los niveles de variabilidad genética entre poblaciones de cóndores andinos a través de un extenso rango geográfico desde Colombia hasta el centro de Argentina y Chile se examinaron secuencias de ADN mitocondrial realizándose el secuenciamiento de la región de control en los dominios II y III junto con parte del gen de la subunidad ribosomal 12S, en 30 especímenes de V. gryphus identificando un total de 5 haplotipos diferentes, encontrando una baja diversidad haplotípica (Hd = 0.64) y una baja variabilidad genética relacionada a mega fauna en peligro de extinción (Hendrickson et al. 2003).
Alcaide et al. (2010) en su estudio de estimación no invasiva de tamaños mínimos poblacionales y variabilidad del complejo mayor de histocompatibilidad (CMH) en el cóndor de los andes, probaron la utilidad de los perfiles del CMH como una huella genética en V. gryphus, para dicho fin realizaron el aislamiento de genes polimórficos y posiblemente funcionales identificando 80 individuos a partir de 110 plumas de muda recolectadas en posaderos mediante análisis de polimorfismos conformacionales de cadena sencilla y de secuenciamiento directo de genes del CMH sumado a la identificación del sexo y evaluaciones de la edad de los especímenes. El estudio proporciono patrones de variación genética relevantes en la respuesta inmune frente a patógenos y demostró el uso potencial de los genes del CMH más allá de su bien conocido papel en ecología evolutiva.
En las últimas décadas el muestreo no invasivo a partir del análisis de vestigios biológicos como pelos, plumas, excrementos, orina o mudas, ha facilitado el estudio de especies raras o elusivas (Restrepo 2010), de difícil monitoreo por su comportamiento. En particular, en las aves la utilización de plumas de muda ha sido muy conveniente para la evaluación de poblaciones.
El presente trabajo tiene el objetivo de evaluar la diversidad genética de cóndores andinos (Vultur gryphus) silvestres y en cautiverio en las regiones de Cusco y Apurímac. Para este fin se caracterizó a los individuos mediante secuenciación de la región control y subunidad ribosomal 12S, y los genes Citocromo Oxidasa subunidad I y NADH deshidrogenasa subunidad dos del ADN mitocondrial extraídos de cálamos de plumas de muda, analizándose los niveles de variabilidad genética intrapoblacional mediante datos moleculares e índices de diversidad genética.
Material y métodos
Área de estudio. - Las plumas de muda fueron colectadas en posaderos y zonas de avistamiento de cóndores, ubicados en zonas alto andinas de los distritos de Cusipata Provincia de Quispicanchis, Mollepata y Limatambo Provincia de Anta, Soraypampa un llano del Distrito de Mollepata, en la Región Cusco y los distritos de Cotabambas, Coyllurqui Provincia de Cotabambas e Ihuayllo y Chalhuanca Provincia de Aymaraes, en la Región Apurímac. Asimismo, se tomaron muestras de plumas en los Zoológicos de la Universidad Nacional de San Antonio Abad del Cusco (UNSAAC), Mundo Andino en Tipón Cusco y el Zoológico de Taraccasa de la ciudad de Abancay en Apurímac (Tabla 1 y Figura 1).
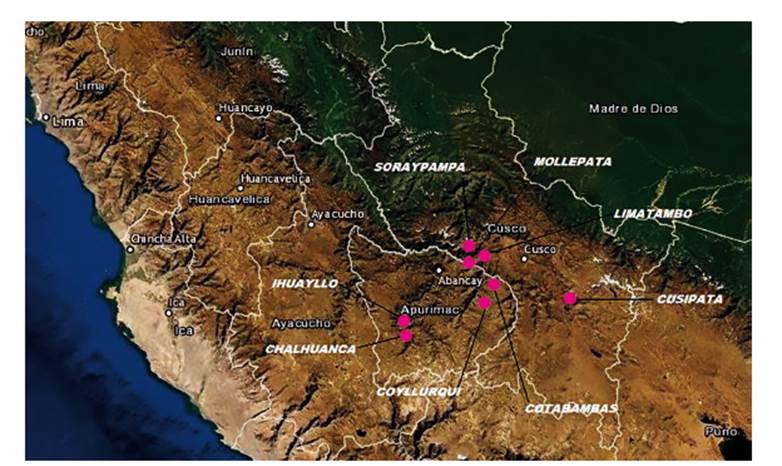
Material biológico. - Para la colecta de plumas se utilizó una técnica de muestreo no invasiva que no requiere la manipulación de los cóndores y posibilita una conservación y almacenamiento simple. En las zonas seleccionadas para el estudio se buscaron plumas de muda frescas (aquellas cuya apariencia física no ha sido afectada significativamente por las condiciones climáticas) y con tamaños mayores a 15 cm. Las plumas fueron colocadas dentro de bolsas de polipropileno estériles con cierre hermético. Se utilizó la diferencia de colores de las plumas para distinguir entre las clases de edad, las aves inmaduras (juveniles y subadultos, hasta 5 - 6 años) presentan plumas marrón grisáceas, mientras que los adultos (> 6 años de edad) tienen plumas de color negro (Wallace & Temple 1987). Cada lugar fue visitado y muestreado solo una vez. Las plumas de zoológicos corresponden a cada individuo en cautiverio.
El procesamiento de las muestras y los diferentes análisis se llevaron a cabo en las instalaciones del Laboratorio de Investigación en Enfermedades Infecciosas del Departamento de Ciencias Celulares y Moleculares de la Universidad Peruana Cayetano Heredia.
Obtención de ADN. - Las muestras de calamos se obtuvieron en condiciones estériles mediante corte y trozado de la parte basal del cañón de las plumas. El ADN se extrajo a partir de 15 muestras de plumas de zoológico y 11 muestras de plumas silvestres. Las muestras fueron reducidas a partículas entre 0.5 - 1mm y digeridas, empleando proteinasa K (20 mg/μL), a 56 °C durante 24 horas. El ADN fue aislado utilizando el kit comercial High Pure PCR Template Preparation, siguiendo las indicaciones del fabricante (Roche Diagnostics, Suiza). El ADN extraído se cuantificó por espectrofotometría empleando el equipo Nanodrop 2000.
Amplificación por PCR. - Se amplificó la secuencia parcial del gen de la región control y la subunidad ribosomal 12S (D-Loop-ARNr12S), secuencia específica para el cóndor andino previamente utilizada por Hendrickson et al. (2003), el gen Citocromo Oxidasa subunidad I (COI) utilizado para código de barras genético y el gen de la NADH deshidrogenasa subunidad 2 (ND2) de amplio uso en evaluaciones de variabilidad genética en poblaciones de aves. La reacción de amplificación se realizó en un volumen final de 25 µL conteniendo buffer PCR 1X, 0.2 mM de dNTPs, 2.5 µM de MgCl2, 0.8 - 1 µM de cada primer, 1.25 unidades de Taq-polimerasa y 3.0 µL de ADN. La amplificación se realizó empleando el termociclador Pro Flex PCR System (Applied Biosystems, Estados Unidos).
La amplificación de la región D-Loop-ARNr12S se realizó empleando los primers L798: 5’-GCAGTTTGCTTTCCATTCG-3’ y H1455: 5’-GGCTGTGCAAGGTGTCTTG-3’ (Hendrickson et al. 2003). La amplificación se basó en un ciclo de desnaturalización inicial a 94 °C por dos min, seguido de 40 ciclos cada ciclo consistente de un paso de desnaturalización a 94 °C por 1 min, alineamiento a 51 °C por 1 min y extensión a 72 °C por 1 min, y un ciclo final de extensión a 72 °C por cinco min.
La amplificación del gen COI se realizó empleando los primers Bird F1: 5’-TTCTCCAACCACAAAGACATTGGCAC-3’ y Bird R1: 5’-ACGTGGGAGATAAT TCCAAATCCTGG-3’ (Hebert 2004). La amplificación consistió en un ciclo de desnaturalización inicial a 94 °C por dos min, cinco ciclos cada ciclo consistente de un paso de desnaturalización a 94 °C por 1 min, alineamiento a 45 °C por 1 min 30 s, extensión a 72 °C por 1 min 30 s, seguido de 30 ciclos cada ciclo consistente de un paso de desnaturalización a 94 °C por 1 min, alineamiento a 51 °C por 1 min 30 s, extensión a 72 °C por 1 min 30 s, y un paso final de extensión a 72 °C por cinco min.
La amplificación del gen ND2 se realizó empleando los primers L5143: 5’-GAACCTACACARAAGRGATCAAAAC- 3’ y H6313: 5’-ACTCTTRTTTAAGGCTTT GAAGGC-3’ (Lerner & Mindell 2005). La amplificación consistió de un ciclo inicial de desnaturalización a 94 °C por 1 min, seguido de 40 ciclos cada uno consistente de un paso de desnaturalización a 95 °C por 1 min, alineamiento a 53 °C por 1 min, extensión a 72 °C por 1 min, y un paso final de extensión a 72 °C por 5 min.
Los productos de amplificación fueron analizados mediante electroforesis en geles de agarosa al 1.5% (p/v) preparados en buffer TAE 1X, teñidos con bromuro de etidio y observados con luz ultravioleta. Los productos de amplificación fueron secuenciados por la empresa Macrogen (Rockville, Maryland, USA. https://www.macrogenusa.com), y están depositados en el GenBank con números de acceso del MW588503 al MW588580.
Los electroferogramas de las secuencias de ADN fueron analizadas empleando el software SeqTRACE (Stucky 2012) y el programa DNA Baser versión 4.36.0.2 (Heracle BioSoft www.DnaBaser.com), luego la identidad de la especie fue corroborada empleando la herramienta BLAST del NCBI (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Posteriormente se realizó el alineamiento múltiple de las secuencias usando el programa Clustal W integrado a Mega 6 (Tamura et al. 2013).
Análisis de datos genéticos. - Se estimó la diversidad haplotípica (Hd) (Nei 1987), la diversidad nucleotídica (π) (Nei & Li 1979), el número promedio de diferencias nucleotídicas (K) (Tajima 1993) y el número de sitios polimórficos, variables o segregantes (S). Asimismo, se estimaron datos moleculares como el número de transiciones (Ts), el número de transversiones (Tv), la composición nucleotídica, el número de sitios parsimoniosamente informativos (es decir se infiere homología filogenética por ancestría común como la explicación más sencilla de la variación) y el número de mutaciones (ɳ). Los parámetros se estimaron con el paquete estadístico Arlequín versión 3.1.1. (http://cmpg.unibe.ch/software/arlequin35/) (Excoffier & Lischer 2010), y el software DnaSP versión 5.1.1 (Librado & Rozas 2009).
Resultados y discusión
Extracción de ADN. - Se obtuvo en promedio una concentración de ADN de 31.22 ng/µL, con un rendimiento promedio de 147.44 ng por mg de muestra para cóndores en cautiverio y una concentración promedio de ADN de 52.38 ng/µL, con un rendimiento de ADN de 339.68 ng por mg de muestra para cóndores silvestres.
Amplificación del ADN mitocondrial. - La amplificación de las tres secuencias mitocondriales (la región control mitocondrial y la subunidad ribosomal 12S, el gen citocromo oxidasa subunidad I y el gen NADH deshidrogenasa subunidad 2) provenientes de muestras de calamos de plumas de cóndores en cautiverio y silvestres, dio como resultado bandas únicas, claras y definidas, con productos de amplificación de 600; 704 y 1090 pb, respectivamente (Figs. 2, 3 y 4).
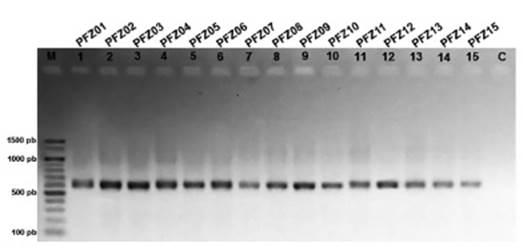
Figura 2 Variabilidad genética del ADN mitocondrial de Vultur gryphus en Cusco y Apurímac a partir de plumas de muda. Productos de amplificación del gen D-Loop-ARNr12S. M: marcador de peso molecular, C: control negativo.
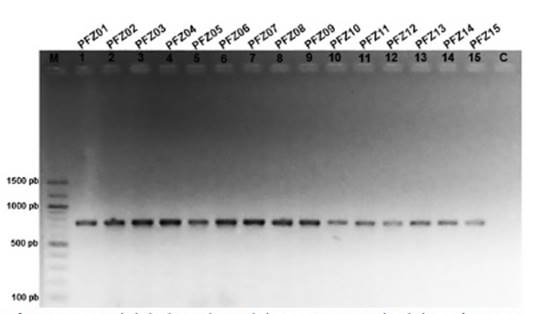
Figura 3 Variabilidad genética del ADN mitocondrial de Vultur gryphus en Cusco y Apurímac a partir de plumas de muda. Productos de amplificación del gen Citocromo Oxidasa subunidad I. M: marcador de peso molecular, C: control negativo.
Figura 4 Variabilidad genética del ADN mitocondrial de Vultur gryphus en Cusco y Apurímac a partir de plumas de muda. Productos de amplificación del gen NADH deshidrogenasa subunidad II. M: marcador de peso molecular, C: control negativo.
Los resultados demuestran que el ADN mitocondrial sobrevive al proceso de queratinización siendo las plumas de muda del cóndor andino una fuente útil y valiosa para el análisis genético mediante PCR. Ademas la amplificación del 100% de los casos constrasta con las afirmaciones de Segelbacker (2002) quien señaló que el éxito de la amplificación es mayor en plumas frescas que en las plumas de muda.
Análisis de la diversidad genética.- La identidad taxonómica molecular de las tres secuencias fue verificada mediante la búsqueda de similitud con las secuencias de referencia en la base de datos del NCBI GenBank, encontrando un 96% de identidad para el segmento D-Loop-ARNr12S y un 98% de identidad para los segmentos COI y ND2 con los genes de V. gryphus de las accesiones AY129646.1, KF446142.1 y KX534724.1 respectivamente. Esto permite comprobar el potencial de los primers utilizados para la identificación y caracterización del cóndor andino.
Las secuencias fueron comparadas encontrando que el segmento D-loop-ARNr 12S presenta una composición nucleotídica promedio con mayor cantidad de A (40.65%) seguido de C (25.87%), T (22.79%) y G (10.68%), en cambio el gen COI presenta mayor cantidad de C (30.83%) seguido de T (27.12%), A (25.57%) y G (16.48%), mientras que el gen ND2 presenta mayor cantidad de C (34.31%), seguido de A (31.46%), T (23.86%) y G (10.37%). Se puede observar que el contenido de A+T es mayor que el de G+C en todos los casos con valores en la matriz general de 57.15% frente a 42.85%, respectivamente. Con relación a las sustituciones nucleotídicas, se encontró que en dos de los genes hay un mayor número de transiciones que de transversiones, así, en el segmento D-Loop-ARNr12S los dos sitios polimórficos encontrados fueron transiciones, en el gen ND2 encontramos tres transiciones y dos transversiones, mientras que en el gen COI hay una transición y cuatro transversiones.
Análisis de sitios polimórficos y cambios nucleotídicos. - El alineamiento de los 600 pb de la región D-loop-ARNr12S reveló cuatro haplotipos (D1, D2, D3, D4), registrándose dos sitios polimórficos en las posiciones 36 y 96 del amplicón, en estos sitios se presentaron dos cambios T - C y dos cambios A - G, es decir que ocurrieron cuatro transiciones. El haplotipo D1 es el más frecuente (73.08%), seguido del haplotipo D3 (19.23%) mientras que los haplotipos D2 y D4 son los menos frecuentes (3.85%). El haplotipo D1 incluye tanto individuos silvestres (n=9) como en cautiverio (n=10) y se encuentra en mayor proporción en los individuos de Cusco, el haplotipo D2 solo se encuentra en Apurímac (n=1), el haplotipo D3 se presenta más en individuos en cautiverio (n=4), mientras que el haplotipo D4 solo se encuentra en un individuo en cautiverio de las localidades de Cusco.
La secuencia del fragmento del gen COI mostró cinco sitios polimórficos, en las posiciones 40, 61, 65, 73 y 605 del amplicón. Se observó un cambio C - A, un cambio C - G, dos cambios A - C, un cambio T - C. El número de haplotipos obtenidos fue de seis (C1 - C6), siendo el haplotipo C1 el más frecuente (73.08%), seguido de los haplotipos C3 y C4 (7.69%) y los haplotipos C2, C5 y C6 los menos frecuentes (3.85%). El análisis de estas secuencias revela que el haplotipo C1 se encuentra más en individuos en cautiverio (n=12) que en los silvestres (n=7), y más abundante en Cusco que en Apurímac. El haplotipo C2 solo está presente en un individuo silvestre de Apurímac, el C3 solo presente en individuos silvestres de Apurímac (n=2), el haplotipo C4 se encuentra tanto en individuos silvestres (n=1) como en cautiverio (n=1), de la región Cusco. El haplotipo C5 se encuentra en un individuo en cautiverio de Cusco y el haplotipo C6 solo en un individuo en cautiverio en Apurímac.
Para el grupo de secuencias del gen ND2 se registran cinco sitios polimórficos en las posiciones 97, 123, 1002, 1050 y 1052 del amplicón. En los cuales se observó dos cambios A - G, un cambio G - T, dos cambios C - T y un cambio A - C, obteniéndose cinco haplotipos (N1 - N5). El haplotipo N1 fue el predominante (84.62%), mientras que los haplotipos N2, N3, N4 y N5 fueron los menos frecuentes (3.85%). El haplotipo N1 fue predominantemente en los individuos en cautiverio (n=13) de la región Cusco, los haplotipos N2 y N4 solo se presentan en individuos en cautiverio de Cusco (n=1), el haplotipo N3 está solo en un individuo silvestre de Cusco y el haplotipo N5 está presente en un individuo silvestre de Apurímac. Cabe destacar que los haplotipos D1, C1 y N1 se presentan con mayor frecuencia en los individuos procedentes de Cusco.
Las regiones estudiadas presentaron escaso número de haplotipos restringidos, solamente el haplotipo C3 es un haplotipo restringido en los cóndores silvestres de Apurímac, mientras que el C4 es un haplotipo restringido a los individuos procedentes de Cusco. Asimismo, el bajo número de haplotipos compartidos indica bajos niveles de flujo génico y es indicio de que no hay expansión poblacional. La distribución en la zona de estudio de los haplotipos D1, presente en todas las localidades de muestreo incluidos todos los zoológicos, C1 presente en todas las localidades con excepción de Mollepata en Cusco e Ihuayllo en Apurímac y N1 presente en todas las localidades con excepción de Cusipata en Cusco, indicaría que en el pasado hubo un alto nivel de flujo génico en las poblaciones de cóndor andino lo cual se evidencia en la actualidad por la alta frecuencia que presentan estos haplotipos.
La Tabla 2 muestra las secuencias variables en genes, edad y localidad de los individuos analizados tanto en cautiverio como silvestres considerando la determinación de la edad por coloración de las plumas y la localidad de las muestras. Tomando en consideración la información genética obtenida de las secuencias de los tres genes utilizados se logró caracterizar 11 genotipos diferentes encontrando que hay tres grupos idénticos, así, el primero agrupa a las muestras PS03 y PS06, el segundo a las muestras PS05, PZ06 y PZ09 y el tercer grupo y el más numeroso agrupa a las muestras PS07, PS09, PS10, PS11, PZ02, PZ04, PZ05, PZ07, PZ08, PZ11, PZ12, PZ13 y PZ14, las demás muestras se encuentran como genotipos únicos. Se observa que el primer grupo esta solo presente en individuos silvestres mientras que en el segundo y tercer grupo se encuentran tanto individuos silvestres como en cautiverio.
Tabla 2 Caracterización de la variación genética de Vultur gryphus en las regiones de Apurímac y Cusco en el sur del Perú.
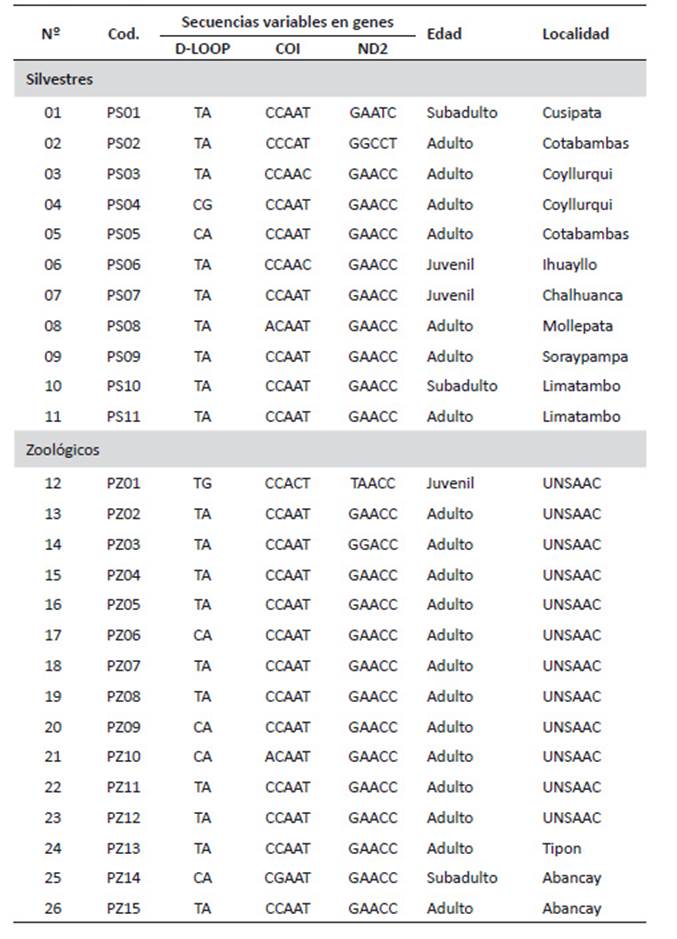
El alineamiento múltiple mostró que el gen COI presentó la porción más variable del ADN analizado permitiendo una evidente diferenciación de las muestras, comprobando que es un material informativo muy conveniente para estudios a nivel poblacional e intraespecífico en el cóndor andino. Examinando la distribución de la variabilidad entre las posiciones de los alineamientos, se encontró que las zonas conservadas se localizan en las posiciones mediales mientras que las zonas con mayor proporción de polimorfismo se localizan en los bordes de los alineamientos.
Diversidad molecular de genes mitocondriales de cóndor andino. - El análisis de las secuencias de la fracción de 600 pb de la región D-Loop-ARNr12S alineadas con la secuencia de referencia AY129646.1 mostró dos sitios parsimoniosamente informativos, dos mutaciones puntuales y dos sitios polimórficos, los cuales fueron 100% transiciones, que definieron cuatro haplotipos. La diversidad haplotípica fue de 0.443, la diversidad nucleotídica de 0.00086, y el número promedio de diferencias nucleotídicas de 0.51692.
Del total de 704 pb correspondientes a la región del gen COI alineadas con la secuencia de referencia KF446142.1 mostró la presencia de dos sitios parsimoniosamente informativos, cinco mutaciones puntuales y cinco sitios polimórficos, de los cuales el 80% fueron transversiones y el 20% fueron transiciones, que revelaron 6 haplotipos, con una diversidad haplotípica de 0.468, una diversidad nucleotídica de 0.00075, y un número promedio de diferencias nucleotídicas de 0.52615.
En base a las secuencias del fragmento de 1090 pb del gen ND2 alineadas con la secuencia de referencia KX534724.1 se encontró la presencia de un sitio parsimoniosamente informativo, cinco mutaciones puntuales y cinco sitios polimórficos, de los cuales el 60% fueron transiciones y el 40% fueron transversiones, que detectaron cinco haplotipos. Además, la diversidad haplotípica fue de 0.289, la diversidad nucleotídica fue de 0.00042, el número promedio de diferencias nucleotídicas fue de 0.45538 (Tabla 3).
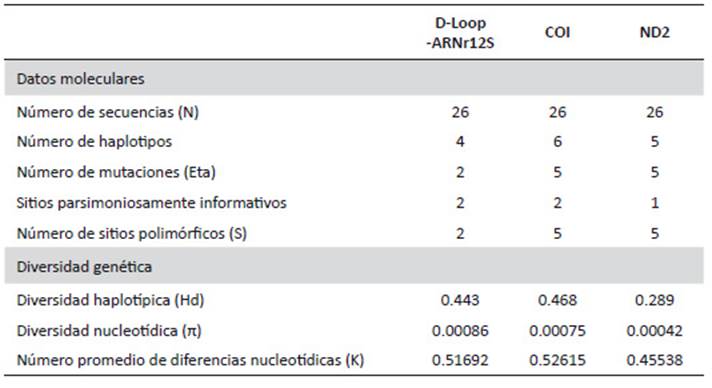
Los valores de diversidad haplotípica encontrados en el presente estudio son muy bajos, lo que sugiere que el total de la muestra de cóndores andinos presenta una baja diversidad genética. Cuando un haplotipo específico es abundante en una población, es de esperar que dicha población presente una baja diversidad haplotípica. También, a menor proporción de lugares polimórficos observados en una población, menor resultará la cantidad de diferencias a pares entre los haplotipos, de este modo siempre será menor la diversidad nucleotídica. Consideramos que un mecanismo que contribuye al descenso de la variabilidad en la población es el reducido tamaño efectivo de dicha población causando esta baja variabilidad mediante procesos genéticos poblacionales convencionales como la pérdida no selectiva de alelos por deriva genética aleatoria tal como lo menciona Crow y Kimura (1970).
Diversidad molecular del cóndor andino por factores evaluados. - De forma complementaria se realizó el análisis de la diversidad genética del cóndor andino en base a los dos factores: condición de vida y ubicación, analizados en forma independiente. Así, al observar los datos moleculares e índices genéticos según el factor condición de vida se lograron evaluar 11 secuencias silvestres y 15 secuencias en cautiverio, encontrando tres haplotipos para el gen D-Loop-ARNr12S, cuatro haplotipos para el gen COI y otros tres haplotipos para el gen ND2 en ambos casos. Asimismo, se presentaron no más de cuatro mutaciones como sitios polimórficos y no más de un sitio parsimoniosamente informativo en ambas condiciones de vida.
En el caso del gen D-Loop-ARNr12S, los cóndores cautivos presentaron mayor diversidad haplotípica (0.514) y; nucleotídica (0.00092), así como un mayor promedio de diferencias nucleotídicas (0.55238) en relación a los cóndores silvestres. En cambio, en el caso de los genes COI y ND2 los cóndores silvestres presentaron mayor diversidad haplotípica, nucleotídica y promedio de diferencias nucleotídicas. Nuestros resultados sugieren que los individuos de cóndores silvestres presentan mayor diversidad genética que los cóndores que se encuentran en cautiverio y para ambas condiciones de vida, encontramos que las estimaciones de diversidad haplotípica, nucleotídica y número promedio de diferencias nucleotídicas son bastante bajos, con intervalos de 0.257 a 0.600 de Hd, 0.00024 a 0.00098 de π y 0.26667 a 0.72727 de K.
El análisis de la diversidad genética del cóndor andino según su ubicación, mostro en los individuos del Cusco la presencia de tres haplotipos en la región D-Loop-ARNr12S y el gen COI y cuatro haplotipos en el gen ND2, mientras que en Apurímac se encontró tres haplotipos en la región D-Loop-ARNr12S, cuatro en el gen COI y dos en el gen ND2.
Los cóndores muestreados en la región Apurímac presentan mayores valores de diversidad haplotípica, nucleotídica y promedio de diferencias nucleotídicas para los tres fragmentos de genes evaluados con valores máximos de 0.750 de Hd, 0.00132 de π y 0.92857 de K para el gen COI. Mientras que en la región Cusco la diversidad haplotípica de los cóndores alcanzó un valor máximo de 0.386, una diversidad nucleotídica máxima de 0.00068 y un valor máximo en el promedio de diferencias nucleotídicas de 0.40523, todos estos valores para el gen D-Loop-ARNr12S.
Para el ADN mitocondrial encontramos que las estimaciones de diversidad haplotípica y de diversidad nucleotídica son menores de 0.5 y 0.0008 respectivamente. Asimismo, el número promedio de diferencias nucleotídicas entre los haplotipos son menores de 0.6 tanto en la estimación de diversidad genética para cada gen, como para cada condición estudiada. Esto indica niveles de variabilidad genética baja en los cóndores analizados, sugiriendo que es una población con estructura genética homogénea que está sufriendo una pérdida del potencial de adaptabilidad lo que supone un riesgo a su sobrevivencia y conservación de la especie en la región sur del Perú.
Esta situación probablemente se deba en parte a que el cóndor andino presenta la característica de ser un ave grande que genera una sola cría, anida una vez cada dos años y no alcanza la madurez sexual hasta los ocho años (Wallace & Temple 1988, Temple & Wallace 1989); sin embargo, es posible también que las diferentes amenazas que viene sufriendo la población de cóndor en el sur del Perú sea un factor que contribuya a la baja variabilidad genética. Asimismo, esta condición estaría implicada en un lento restablecimiento de su situación de cuello de botella a causa de la baja población evidenciado por la baja variabilidad genética que presenta. Así, la masa corporal grande y la reducción del índice metabólico asociado podrían retrasar el almacenamiento de nuevas variantes genéticas disminuyendo los índices de mutación (Bleiweiss 1998, Mindell et al. 1996, Martin & Palumbi 1993). También mutaciones nuevas se esparcirían en forma lenta a través de una población escasamente distribuida y los fenómenos alternantes propios de las mitocondrias podrían disminuir la variabilidad por uno o más mecanismos.
Consideraciones sobre la variabilidad genética y conservación en el cóndor andino.- La baja variabilidad genética encontrada en los cóndores en el presente estudio, se puede interpretar como la presencia de sensibilidad de este organismo a la posible declinación de su población en Cusco y Apurímac y por ende encontrar un estado de vulnerabilidad en esta especie. Si bien no se cuenta con estudios poblacionales detallados a nivel nacional, se estima que la población en el Perú viene disminuyendo, situación que la ubica actualmente en la categoría de En Peligro (EN) (Piana 2018). Esto acarearía efectos graves en la eficacia biológica desde la reducción de la fertilidad, la endogamia hasta un incremento en la probabilidad de extinción (Lacy 1997).
El conocimiento de la diversidad genética de las poblaciones de V. gryphus proporciona una base científica y una herramienta adicional para poder desarrollar políticas de ordenamiento que posibiliten la protección de los ambientes en los cuales se desarrolla esta especie considerando su longevidad y madurez tardía; asimismo, aplicar la legislación nacional y efectuar acuerdos de cooperación internacional debido a las migraciones que realiza con el fin de disminuir la declinación de la población actual.
En el contexto de la planificación de proyectos que consideren medidas de conservación in situ y ex situ, sería importante: localizar las zonas relevantes para la especie a través del mapeo de nidos, dormideros y posaderos, definir la existencia efectiva del alimento considerando a los competidores para establecer la cantidad conveniente de cóndores que posibiliten salvaguardar el potencial reproductivo en cada región, evaluar parámetros poblacionales precisando los factores de mortalidad e índices de supervivencia por edad y región, otro aspecto importante es la urgente necesidad de la realización de un censo a nivel nacional, que permita conocer la situación de las poblacionales actuales.

