










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkAnales de la Facultad de Medicina
versão impressa ISSN 1025-5583
An. Fac. med. v.67 n.3 Lima jul.- sep. 2006
Demencia asociada a infección por virus de inmunodeficiencia humana tipo 1
Nilton Custodio 1, Juan Escobar 2, Juan Altamirano 3
1 Unidad de Diagnóstico de Deterioro Cognitivo y Prevención de Demencia-Servicio de Neurología. Clínica Internacional. Lima, Perú.
2 Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú
3 Servicio de Neurología. Clínica Angloamericana. Lima, Perú.
Resumen
La demencia asociada a infección por virus de inmunodeficiencia humana (DVIH) es una entidad caracterizada por la tríada de compromiso cognitivo, síntomas conductuales y motores, que generan serias dificultades en la capacidad funcional del paciente. Las múltiples denominaciones generan confusión y alta probabilidad de subreconocimiento. No obstante, la incidencia de DVIH es controversial; se tiene claro que más de 90% de pacientes con sida tiene anormalidades neuropatólogicas compatibles con DVIH. Los mecanismos patogénicos involucran una compleja interacción entre el VIH y las células del cerebro, que generan claramente dos vías incluyentes, la inflamatoria y la no inflamatoria, las cuales generan factores neurotóxicos y quimiotácticos, inductores de apoptosis, que conducen a una disrupción neuronal-glial, probablemente responsable de la injuria y/o muerte neuronal, que conduciría a un fenómeno de neurodegeneración acelerada. Los síntomas son de una demencia subcortical, siendo los síntomas de presentación más comunes el compromiso de la memoria, enlentecimiento mental, dificultad para la marcha y depresión. El diagnóstico es esencialmente clínico y se realiza por exclusión. Son de utilidad práctica la HIV Dementia Scale (HDS) y la International HIV Dementia Scale (IHDS), como pruebas iniciales de descarte. El tratamiento debe incluir la combinación de antiretrovirales y neuroprotectores. Como conclusión, la DVIH es una complicación devastadora de la infección por VIH que debe ser reconocida tempranamente.
Palabras clave: Demencia; HIV-1; síndrome de inmunodeficiencia adquirida.
Human immunodeficiency virus type 1 infection associated dementia
Abstract
Dementia associated to human immunodeficiency virus infection (DHIV) is an entity distinguished by three main signs -cognitive, behavioral and motor symptoms- which generate serious difficulties in the functional capacity of the patient. The multiple denominations generate confusion and diagnostic difficulties. In spite of controversy in DHIV incidence, it is clear that more than 90% of patients with AIDS has compatible neuropathological anormalities with DHIV. The pathogenic mechanisms involve complex interactions between the HIV and the brain cells generating two inclusive paths, inflammatory and non inflammatory, that produce neurotoxic and chemotactic factors, inductors of apoptosis that lead to neuro-glial disruption probably responsible of injury and/or neuron cell death, that finally would lead to accelerated neurodegeneration phenomenon. Symptoms are subcortical dementia, mental sluggishness, walking difficulties and depression. Diagnosis is essentially clinical and by exclusion. The HIV Dementia Scale (HDS) and the International HIVD Scale (IHDS) are of practical usefulness as initial screening tests. Treatment should include the combination of antiretrovirals and neuroprotectors. We conclude that DHIV is a devastating complication of HIV infection that should have early recognition.
Key words: Dementia; HIV-1; acquired inmunodeficiency syndrome.
INTRODUCCIÓN
Diferentes desórdenes neurológicos han sido reconocidos como complicaciones del virus de la inmunodeficiencia humana tipo 1 (VIH-1), desde los inicios de la epidemia del síndrome de inmunodeficiencia adquirida (Sida) (1). Estas complicaciones ocurren en cualquier estadio de la infección y pueden comprometer el sistema nervioso central y/o el sistema nervioso periférico (2-5). Aproximadamente, 10% de pacientes tiene síntomas neurológicos (6,7), 30 a 50% desarrollan complicaciones neurológicas durante el curso de la enfermedad (1,6) y más de 90% de pacientes muestra evidencia patológica de enfermedad neurológica, durante la necropsia (8-10). Estas complicaciones neurológicas se pueden presentar como resultado de infecciones oportunistas y neoplasias (por compromiso de la inmunidad celular) o ser el resultado del efecto directo del VIH-1 sobre el sistema nervioso, entre las que se incluye demencia, mielopatía, miopatía y polineuropatía sensorial distal (2). La tríada de declinación cognitiva, cambios conductuales y disfunción motora es denominada demencia asociada a infección por VIH-1 en adultos y encefalopatía por VIH-1 en niños; es una de las más devastadoras y enigmáticas complicaciones neurológicas primarias del VIH-1 (2,11).
Las anormalidades cognitivas han sido reconocidas desde los inicios de la epidemia del sida y ha recibido múltiples denominaciones a través del tiempo: encefalitis subaguda (12,13), encefalitis a células gigantes multinucleadas (2,11), encefalopatía VIH-1 (12-14), desorden cognitivo/motor asociado a infección por VIH-1 (2,4,12,13,15), complejo demencia sida (4,11,13,15), demencia sida (15-17) y, recientemente, demencia asociada a infección por VIH-1 (DVIH) (5,11,13,15,18,19). Los términos desorden cognitivo/motor asociado a infección por VIH-1 y complejo demencia sida fueron usados frecuentemente como sinónimos, pero el Working Group of the American Academy of Neurology Adquired Inmune Deficiency Syndrome Task Force las caracterizó como dos categorías clínicas progresivas del mismo proceso, según la severidad del compromiso de las actividades de la vida diaria. La forma menos severa fue denominada desorden cognitivo/motor menor (DCMm) asociado a infección por VIH-1, y la forma más severa fue denominada complejo demencia sida (12) o desorden cognitivo/motor mayor (DCMM) asociado a infección por VIH-1, hoy conocida como DVIH. En 1986, el término complejo demencia sida fue introducido para incluir pacientes con deterioro cognitivo asociado a mielopatía (20). Sin embargo, en la actualidad, a pesar que el término enfatiza que el trastorno cognitivo es la afección predominante, también implica que no es la única manifestación clínica (15). Está claro que demencia y mielopatía son enfermedades separadas en el contexto del sida y que los pacientes pueden ser afectados por uno o ambos procesos a la vez; por lo que, es un término que cada vez debe ser menos empleado. En 1987, el término encefalopatía VIH, hoy conocida como DVIH, fue adicionado a la lista de enfermedades que definen la condición de sida (21). Desde entonces, DVIH ha sido estimado como la manifestación inicial de sida en 0,8% a 5,5% de pacientes. En el presente artículo, vamos a resumir la epidemiología, características clínicas, patogénesis y tratamiento de la DVIH.
EPIDEMIOLOGÍA
La incidencia exacta de DVIH en individuos infectados con VIH-1 es desconocida. Sin embargo, aproximadamente en 30% de pacientes con sida en estadios avanzados se desarrolla trastorno cognitivo (22). Los cuadros de demencia se desarrollan en 15% a 20% de pacientes, con una incidencia anual de 7%, después del reconocimiento de la condición de sida (20). Se estima que la ocurrencia actual puede ser mayor, debido a que muchos casos de DVIH pueden no ser diagnosticados cuando otra enfermedad que pone en riesgo la vida del paciente está presente. De hecho, algunas series de autopsias muestran anormalidades neuropatológicas en más de 90% de pacientes con sida (8-10). Por otro lado, estudios rutinarios de autopsias sugieren que la neuropatología del VIH-1 y DVIH no se ha alterado en los últimos 15 años, y el cerebro continúa siendo el segundo órgano más afectado (23,24), a pesar de los claros efectos benéficos de la moderna terapia de combinación retroviral o terapia antiretroviral de gran actividad (Targa).
No obstante que la DVIH es ocasionalmente una enfermedad que define sida (2,20), ésta usualmente se desarrolla tardíamente, en pacientes con inmunosupresión severa, cuando el recuento de linfocitos CD4 es menor de 200 pmmc (11). Otros factores que pueden incrementar el riesgo de anormalidades neuropsicológicas en forma temprana son controversiales. Así, algunos investigadores han sugerido que los usuarios de drogas intravenosas (25,26) tienen un mayor riesgo de desarrollar DVIH, que aquellos individuos con otras conductas de riesgo; pero, estos hallazgos no han sido reproducidos por otros autores (27,28). Debido a la asociación encontrada entre nivel de educación bajo y demencia de tipo Alzheimer, se viene otorgando especial atención a la probable asociación entre este factor de riesgo y el trastorno cognitivo en pacientes con infección VIH-1. Existen estudios (29-32) que reportan una elevada asociación entre niveles de educación bajos y compromiso cognitivo asociado a infección VIH-1. Dos de estos estudios (30,32) incluyen varones homosexuales asintomáticos, de clase media, con buena educación, y uno (29) incluyó individuos asintomáticos usuarios de drogas endovenosas. Otro estudio (31) incluyó individuos con diferentes conductas de riesgo, diferente severidad de la enfermedad y varios niveles educacionales. Un estudio de casos y controles, realizado en individuos consecutivos seronegativos, seropositivos asintomáticos y seropositivos sintomáticos, demostró asociación entre niveles de educación bajos, disminución de niveles de CD4 y conductas de riesgo homosexual/bisexual y heterosexual, con mayor riesgo de desarrollar trastorno cognitivo en individuos seropositivos con infección VIH-1 (33). Sin embargo, dichos estudios han sido realizados en países desarrollados, donde el factor educación puede ser un indicador de diferentes factores.
Aparte de la educación, otros factores han sido esporádicamente asociados con elevada probabilidad de ocurrencia de trastorno cognitivo asociado a infección VIH: mayor edad (20), sexo femenino, disminución de los niveles de hemoglobina (20,34), menor índice de masa corporal (20), deficiencia de vitamina B12 (35), injuria cerebral traumática previa (36) y síntomas depresivos concurrentes (33). Según una investigación de Valcour, en una cohorte Hawaiana de pacientes con VIH-1, la DVIH es más frecuente en individuos seropositivos mayores de 50 años de edad, comparado con seropositivos de 20 a 39 años de edad (25,2% vs. 13,7%, p=0,041) (37). En la misma cohorte, se ha demostrado, después de controlar posibles factores de riesgo, como edad y diabetes, que la apolipoproteína E4 (APOE4) es un factor de riesgo independiente para DVIH en individuos seropositivos mayores de 50 años de edad, comparados con seropositivos de 20 a 39 años de edad (38).
CARACTERÍSTICAS CLÍNICAS DE DVIH
La DVIH usualmente es un proceso subagudo, cuyas características sugieren una demencia de tipo subcortical. La DVIH es un diagnóstico de exclusión (2), con síntomas y signos tempranos que suelen ser difíciles de reconocer (11), por lo que es necesario tener un adecuado plan diagnóstico. Se agrupa los síntomas clínicos en tres categorías: cognitivos, conductuales y motores (5,11). Los síntomas cognitivos incluyen compromiso de la memoria de corto plazo, pobre concentración, enlentecimiento mental y pérdida de la flexibilidad y espontaneidad. Cambios de personalidad, apatía, irritabilidad y depresión caracterizan los cambios conductuales. Los síntomas motores pueden ser evidenciados como torpeza o enlentecimiento motor fino, tremor y debilidad de miembros inferiores (11,15). En una serie de 299 pacientes con DVIH, evaluados en la Universidad Johns Hopkins, se mostró que el compromiso de la memoria, enlentecimiento mental, dificultad para la marcha y depresión fueron los 4 síntomas de presentación más comunes (39). Raramente, el desorden puede iniciarse abruptamente y progresar rápidamente; siendo 6 meses el tiempo promedio de sobrevida, una vez realizado el diagnóstico, sí el paciente no recibe tratamiento.
Según MacArthur, después de la era Targa, se han producido cambios en el fenotipo y la progresión temporal en la DVIH y plantea tres distintos subtipos: (1) Demencia subaguda progresiva, en pacientes no tratados con un síndrome clínico de demencia severa progresiva, similar a la observada en la era pre-Targa; (2) Demencia crónica activa, en pacientes que reciben Targa con pobre adherencia o con resistencia viral, y están en riesgo de progresión del compromiso neurológico; y, (3) Demencia crónica inactiva, en pacientes bajo tratamiento con Targa con buena adherencia y supresión viral efectiva, quienes permanecen neurológicamente estables (40).
DIAGNÓSTICO DE DVIH
Los test neuropsicológicos son importantes en el diagnóstico de DVIH, pero ellos consumen tiempo y son dependientes del nivel de educación y lenguaje del evaluado, pero sobre todo, no son disponibles en países en desarrollo. Así, instrumentos de evaluación breves han sido desarrollados para detectar síndromes específicos de demencia, como por ejemplo el Mini Mental State Examination (MMSE); sin embargo, éste fue diseñado para detectar demencias corticales, como enfermedad de Alzheimer; siendo menos sensible para detectar demencias subcorticales, como la DVIH. La HIV Dementia Scale (HDS) es un test breve pero sensible para detectar pacientes seropositivos en riesgo de desarrollar DVIH (41) y evalúa rapidez motora, memoria, praxis constructiva y funciones ejecutivas (Tabla 1). No obstante, ha demostrado ser sensible y bien tolerado en pacientes con DVIH y demencia vascular. También, se ha demostrado cierta dificultad en su administración cuando es aplicado por médicos no neurólogos. Recientemente, Sacktor y colaboradores han evaluado la International HIV Dementia Scale (IHDS) en una cohorte de individuos de Estados Unidos y Uganda, demostrando ser una herramienta útil para identificar individuos con VIH-1 en riesgo para desarrollar DVIH, tanto en países industrializados como en países en vías de desarrollo (42). Este test evalúa rapidez motora, rapidez psicomotora y recuerdo de 4 palabras a los 2 minutos (Tabla 2).


Para su aplicación, se inicia con el registro de las siguientes cuatro palabras: perro, abrigo, frejol y rojo. Para evaluar la rapidez motora, se pide al evaluado que realice una simulación de pinza con su primer y segundo dedo de la mano no dominante, tan amplio y rápido como sea posible. Para evaluar la rapidez psicomotora, se pide al evaluado que realice movimientos secuenciales y sucesivos de la mano no dominante, tan rápido como sea posible; según el siguiente orden: primero, mano extendida y palma hacia arriba, sobre una superficie plana (el escritorio o la mesa de la evaluación); segundo, mano extendida y palma hacia abajo sobre la misma superficie plana; y, tercero, mano extendida, perpendicular a la misma superficie plana, con base en el quinto dedo. Para la evaluación del recuerdo de 4 palabras, se solicita al paciente que diga las 4 palabras que registró al inicio del test. En esta parte final del test, es posible ayudar al evaluado con claves semánticas. Para la puntuación total del test, se procede de la siguiente manera:
Rapidez motora: Puntaje máximo de 4
4 = 15 pinzas en 5 segundos
3 = 11 a 14 pinzas en 5 segundos
2 = 7 a 10 pinzas en 5 segundos
1 = 3 a 6 pinzas en 5 segundos
0 = 0 a 2 pinzas en 5 segundos
Rapidez psicomotora: Puntaje máximo de 4
4 = 4 secuencias en 10 segundos
3 = 3 secuencias en 10 segundos
2 = 2 secuencias en 10 segundos
1 = 1 secuencia en 10 segundos
0 = no puede realizar la prueba
Memoria-Recuerdo: Puntaje máximo de 4
Un punto por cada palabra respondida espontáneamente.
Medio punto por cada palabra, si requirió ayuda.
El puntaje total de la IHDS se obtiene al sumar los resultados de los tres subtests, siendo este de 12 puntos. Un paciente con puntaje menor o igual a 10 debe ser evaluado por posible demencia.
Por otro lado, cuando se trata de establecer el grado o estadio de demencia, tenemos que referirnos a los dos sistemas de estadios propuestos por Sacktor, en el año 2000 (43), y por Marder, en el año 2003 (44). Con la finalidad de establecer un vocabulario común para estudios epidemiológicos, de historia natural y tratamiento, basados en el examen clínico y la evaluación neuropsicológica, sin tener que recurrir al valioso aporte de los especialistas en neuropsicología, en 1988, se diseñó un sistema de estadios, denominado Memorial Sloan Kettering (MSK) (43) de pacientes con DVIH, en ese entonces, denominada complejo demencia sida, y fue usado en diversos ensayos clínicos y para un estudio de cohortes de individuos con riesgo alto de desarrollar DVIH, por el Dana Consortium on Therapy for HIV Dementia and Related Cognitive Disorders, establecido en febrero de 1994 (45). Sin embargo, esta escala está referida al grado de compromiso funcional en las actividades de la vida diaria; mientras que, detalles de las anormalidades neurológicas y neuropsicológicas no son especificadas; por lo que, en febrero de 1998, el Northeast AIDS Dementia (NEAD) Consortium fue creado para reclutar una cohorte de individuos con riesgo alto de desarrollar DVIH y modificar el sistema de estadios MSK, incluyendo información del examen neurológico y neuropsicológico, así como la evaluación funcional, cuyos resultados iniciales han sido recientemente publicados, el cual ha sido denominado sistema de estadios MSK modificado (MSKm) (44) (Tabla 3).
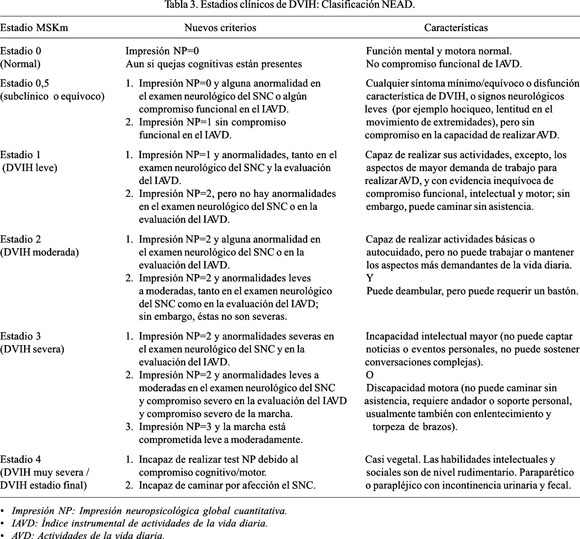
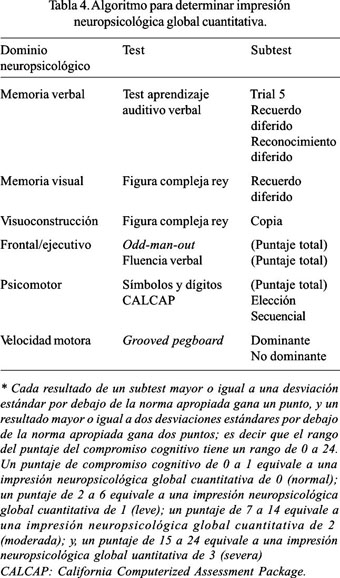
Los criterios de inclusión para DVIH fueron pacientes con infección por VIH-1 y recuento de linfocitos CD4 menor de 200 pmmc o recuento de linfocitos de CD4 menor de 300 pmmc, pero con compromiso cognitivo en alguna prueba neuropsicológica. Los criterios de exclusión fueron pacientes con infección del sistema nervioso central actual, historia clara de esquizofrenia, desorden neurológico compatible con esclerosis múltiple y alguna otra condición que soporte el diagnóstico de demencia tratable (43-45). El examen neurológico debe ser realizado en base a las recomendaciones de AIDS Clinical Trials Group y la subescala motora (part III) de la Unified Parkinson Disease Rating Scale (UPDRS) (46). El examen neuropsicológico debe incluir la evaluación de seis dominios cognitivos: la memoria verbal, mediante el test de aprendizaje auditivo-verbal de Rey (42); la memoria visual, mediante el test de recuerdo diferido, con la figura compleja de Rey-Osterrieth (42); las habilidades constructivas, mediante el test de recuerdo inmediato con la figura compleja de Rey-Osterrieth (47); las habilidades psicomotoras, mediante el test de símbolos y dígitos (48) y el test de reacción (49); las habilidades motoras, mediante el test Grooved Pegboard (50); y el sistema frontal, mediante el test de fluencia verbal (51) y el test Odd-Man-Out (52). De tal manera que, como resultado de esta detallada evaluación se pueda establecer un algorritmo para la determinación de la impresión global cuantitativa de la función cognitiva. Así, con la evaluación de 12 subtests se genera el puntaje de la evaluación neuropsicológica global, de tal manera que, cada resultado de un subtest mayor o igual a una desviación estándar por debajo de la norma apropiada gana un punto, y un resultado mayor o igual a dos desviaciones estándares por debajo de la norma apropiada gana dos puntos; es decir que, el rango del puntaje del compromiso cognitivo tiene un rango de 0 a 24. Un puntaje de compromiso cognitivo de 0 a 1 equivale a una impresión neuropsicológica global cuantitativa de 0 (normal); un puntaje de 2 a 6, equivale a una impresión neuropsicológica global cuantitativa de 1 (leve); un puntaje de 7 a 14 equivale a una impresión neuropsicológica global cuantitativa de 2 (moderada); y, un puntaje de 15 a 24 equivale a una impresión neuropsicológica global cuantitativa de 3 (severa) (44) (Tabla 4). Las mediciones funcionales recomendadas son el Índice Instrumental de Actividades de la Vida Diaria, de Lawton y Brody (53), y la Escala de Automantenimiento Personal, de Living/Lawton, sobre las actividades instrumentales diarias (54). El inventario de depresión de Beck (55) debe ser administrado para evaluar síntomas depresivos. Los ítems de depresión no son incluidos en MSKm, pero ayudan a formar la impresión en la evaluación global cognitiva.
Con todo ello, vale decir que, con la evaluación neurológica, la evaluación neuropsicológica y la medición funcional de las actividades de la vida diaria es posible establecer un diagnóstico correcto y categorización de la DVIH, en base al sistema de estadios MSKm, propuestos por el Consorcio NEAD (44), que pueden ser estadio 0 (normal), estadio 0,5 (subclínico o equívoco), estadio 1 (DVIH leve), estadio 2 (DVIH moderada), estadio 3 (DVIH severa) y estadio 4 (DVIH muy severa) (Tabla 3).
A pesar que la DVIH no puede ser diagnosticada basada en resultados de exámenes de laboratorio y exámenes radiográficos, dichos estudios ayudan a excluir diagnósticos alternativos. Por ejemplo, los estudios de sangre y líquido cefalorraquídeo (LCR) descartan algún proceso infeccioso, capaz de producir un cuadro similar a demencia, como neurosífilis o citomegalovirus; y, además, pueden excluir trastornos metabólicos, como encefalopatía urémica, hepática, trastorno hidroelectrolítico o trastorno en los niveles de glicemia; inclusive, descarta la posibilidad de abuso de alcohol, drogas y efectos colaterales de medicación (3-5,11). Las anormalidades en LCR frecuentemente incluyen pleocitosis mononuclear leve (usualmente < 50 células pmmc, observado en la quinta parte de pacientes), fracción inmunoglobulina total elevada, concentración total de proteínas elevadas (usualmente < 200 mg/dL, observado en dos tercios de pacientes), bandas oligoclonales y síntesis intratecal de anticuerpos IgG contra VIH-1 (3,5,11). En estadios finales de la enfermedad, los marcadores de activación inmune en LCR, como b2- microglobulina, neopterina y quinolinato, generalmente están elevados (2,5,11). Por otro lado, la detección de antígeno p24 de VIH-1 en LCR, independiente de su concentración en suero, correlaciona directamente con DVIH (56). Las neuroimágenes, es decir tomografía cerebral (TC) e imagen por resonancia magnética (IRM) son necesarias para excluir otras infecciones o neoplasias, así como para identificar la atrofia cerebral y los cambios en la sustancia blanca. Las características neuroradiológicas primarias de la DVIH son atrofia cerebral global, dilatación ventricular proporcional y simétrica, anormalidades simétricas de la sustancia blanca (leucoencefalopatía) y mineralización vacuolar en niños (3,11). Existe una correlación entre la cantidad de atrofia cerebral observada en la IRM y la severidad de la demencia (57). Además, se ha demostrado que existe correlación entre la cuantificación del volumen parenquimal cerebral, medido por IRM, con el compromiso neuropsicológico y la disfunción cognitiva motora, en pacientes con infección VIH (58). La relación de los cambios celulares en el cerebro de pacientes afectados por DVIH con el inicio y la progresión de la enfermedad aún no son claros. En estudios de espectroscopia por resonancia magnética (ERM), evaluando el patrón metabólico en individuos seronegativos, seropositivos pero neuroasintomáticos, y en individuos con DVIH, colocando el voxel a nivel de ganglios basales, centro semioval y corteza parietal, se ha identificado los cambios metabólicos regionales. Así, comparado con seronegativos, los neuroasintomáticos tienen marcadores gliales elevados (MI/Cr: proporción de mio-inositol y creatina) en la sustancia blanca, mientras que, en DVIH, se observa además de aumento de MI/Cr en sustancia blanca y ganglios basales, un aumento de Cho/Cr (proporción de colina y creatina) en sustancia blanca y ganglios basales.
Comparado con neuroasintomáticos, en los pacientes con DVIH se observa una reducción de marcador neuronal (NAA/Cr: proporción de N-acetil-aspartato y creatina) en la sustancia blanca frontal, inicialmente, y luego en ganglios basales. Las anormalidades corticales son raras, hasta que la demencia está avanzada (59). En ese sentido, se ha planteado, que la elevación de MI o Cho, asociado con índices microvasculares elevados son consistentes con inflamación, y que la ausencia de una reducción significativa de NAA podría reflejar un proceso eminentemente inflamatorio. Por el contrario, una disminución de NAA, en ausencia de incremento significativo de Cho o MI, podría reflejar incremento de la actividad no inflamatoria, es decir los fenómenos de neurotoxicidad (60).
NEUROPATOGÉNESIS DE DVIH
El virus de inmunodeficiencia humana es un virus ARN, miembro de la subfamilia de los lentivirus, que a su vez pertenece a la familia de los retrovirus (61). El sistema nervioso central (SNC) del ser humano es susceptible a la infección por parte varios tipos de lentivirus, dentro de los cuales se incluye al VIH-1. Los requerimientos específicos para que el VIH ingrese al SNC y a los diferentes tipos celulares que lo resguardan indican una compleja interacción entre el VIH y las células del cerebro. Esta neuroinvasión ocurre de manera temprana (en las primeras semanas), en el curso de la infección del individuo, razón por la cual se postula al SNC como un reservorio viral a lo largo del curso de la enfermedad, debido a que prácticamente provoca un secuestro viral (62-65). En líneas generales, para que el VIH ingrese a cualquier célula del organismo tiene que unirse, fundamentalmente, a la molécula CD4 (que se encuentra en los linfocitos T, monocitos y células dendríticas) y a una familia de correceptores alfa y beta de citoquinas. Estos correceptores son específicos, dependiendo del tipo celular; así, para las proteínas inflamatorias del macrófago 1 alfa y 1 beta (células que conforman la principal línea de defensa del SNC); el principal correceptor utilizado es el CCR5; mientras que, el CXRC4 es el correceptor utilizado por el VIH aislado en la periferia, particularmente en los estadios terminales del sida (63,65-67). La neuroinvasión se realiza a través de la barrera hematoencefálica (BHE), mediante macrófagos infectados, y podría ser potenciada por proteínas virales (fundamentalmente la proteína Tat), que inducen la expresión de genes de moléculas de adhesión y productoras de citoquinas en astrocitos y microglía. Otras formas de invasión del VIH al SNC podrían incluir la infiltración de células infectadas a través de los plexos coroideos y/o a través de una BHE previamente dañada, como ocurre en los consumidores crónicos de alcohol (63,68,69). Algo importante y que da sustento al postulado que el SNC es un reservorio viral es que, a pesar que la penetración del VIH se da en los inicios de la enfermedad, generalmente las deficiencias neurológicas significativas ocurren mucho después, en contraste con las encefalitis virales clásicas (como en la infección por herpes simple y rabia), por lo cual se postula que la lesión del SNC por el VIH sigue una vía indirecta de infección; con lo cual, la disfunción y la muerte neuronal resultarían de eventos que son secundarios a la infección de fagocitos mononucleares, mediada por receptores de citoquinas en conjunción con la molécula CD4 mencionados líneas arriba. Debido a esta afectación indirecta del SNC por el VIH es que se postula 2 vías incluyentes del daño neuronal, una inflamatoria y una no inflamatoria. En este contexto, se define inflamación como la infiltración de células mononucleares y proliferación y activación de células gliales. En contraste, la vía no inflamatoria se basa en la interrupción de la producción de los factores tróficos esenciales para la homeostasis neuronal, como resultado de la infección y lesión de la microglía y el astrocito, aunado a la liberación por parte del VIH de virotoxinas, como las proteínas Tat, Nef y gp120 (60,63,70-72). Aquellas proteínas son directamente neurotóxicas y pueden bloquear el consumo de glutamato por el astrocito, activando la vía del ácido araquidónico e iniciando así una cascada neurotóxica relacionada a destrucción de membranas celulares, mediante la liberación de radicales de oxígeno (estrés oxidativo), que finalmente condiciona la injuria y/o muerte neuronal (72,73). La vía inflamatoria se inicia con la entrada de macrófagos infectados activos vía transporte tipo caballo de Troya (el virus se camufla en los macrófagos para poder ingresar al SNC) y con la activación inmune de la microglía (60,63-65). En ambos procesos, se produce la liberación de factores neurotóxicos, tales como el oxido nítrico (ON), factor del necrosis tumoral alfa (TNF-α), la interleuquina 1 (IL-1), interleuquina 6 (IL-6) y factores quimiotácticos, como la proteína quimioatrayente del monocito tipo I; los cuales van ha inducir apoptosis en la neurona, mediante la activación de caspasas, una familia de proteasas involucradas en la transducción de estímulos apoptóticos y que ordenan el desensamblaje celular. Las caspasas son sintetizadas como proenzimas y son activadas por clivaje proteolítico. Múltiples caspasas pueden activar otras caspasas en forma de una cascada secuencial, que finalmente conducen a la muerte celular; siendo la caspasa-3 el efector de la cascada proteolítica activada en un gran número de desórdenes neurodegenerativos (60,63,70,74). Dichos eventos se dan en términos de un fenómeno de golpe y fuga, que dan como resultado la conformación de un circuito de retroalimentación positiva que conlleva a la autoperpetuación de dicha cascada, que finalmente conduce a una disrupción neuronal-glial, la cual probablemente es la responsable de la injuria y/o muerte neuronal, que conduciría a un fenómeno de neurodegeneración acelerada relacionada a los casos de demencia presentados en las personas con infección por el VIH (60,63,65,73).
TRATAMIENTO DE DVIH
La delineación de la secuencia de pasos fisiopatológicos que contribuyen a la DVIH nos deja claro que, tan solo la terapia antirretroviral no es suficiente para prevenir la activación autosostenida de macrófagos, con la subsecuente liberación de factores neurotóxicos (60); por lo que, se hace necesaria la adición de neuroprotectores.
No obstante, se tiene claro que la monoterapia con altas dosis de zidovudina (AZT) reduce la frecuencia/incidencia de DVIH, son los regímenes de más de tres drogas antirretrovirales considerados los de elección (40). El régimen Targa óptimo para el tratamiento de DVIH aún no ha sido establecido. Inicialmente; se había anticipado, basado en las propiedades farmacocinéticas de las drogas, que podría haber una relación directa de la penetración de la droga al SNC, con mayor eficacia. Sin embargo, a partir de la cohorte MACS, se demostró que la mejoría neurocognitiva con Targa fue independiente de la penetración teórica de las drogas al SNC (75). En ese mismo sentido, los estudios de adición de abacavir -un nuevo inhibidor de la transcriptasa reversa- a Targa no han demostrado disminución adicional en la carga de ARN de VIH-1 en LCR, ni en la performance neuropsicológica (76). Así, hoy en plena era Targa, muchos pacientes con DVIH tratados con Targa permanecen neurológicamente estables por varios años después de haber iniciado Targa o pueden mostrar una reversión parcial de las deficiencias neurológicas (77,78). Todo indica que, las deficiencias cognitivas en individuos con VIH en Targa no permanecen estáticas. De hecho, en el estudio NEAD, se observó una alta frecuencia de transiciones entre los diversos estados neurológicos (normal/DCMm/DVIH) (79). El 55% de los sujetos del estudio mostró una transición, dentro de los cuales, 55% mejoró y 46% empeoró.
Por otro lado, respecto al tratamiento neuroprotector, algunos agentes, como los bloqueadores de canales de calcio, antagonistas NMDA, antagonistas FNT alfa e inhibidores de la producción de radicales libres de oxígeno parecen bloquear potencialmente las vías que conducen a injuria neuroaxonal. El primer ensayo clínico usando un agente neuroprotector para DVIH, fue un ensayo de fase II con nimodipino, el cual mostró una tendencia a la mejoría en las evaluaciones neuropsicológicas con dosis altas (60 mg por vía oral, 5 veces por día), después de 16 semanas de tratamiento (80). Selegilina es un inhibidor irreversible de la monoamino oxidasa, una enzima intracelular asociada con la membrana externa de la mitocondria, que puede tener un efecto neuroprotector, al disminuir la producción de radicales libres de oxígeno. Dos pequeños ensayos clínicos mostraron mejoría significativa en aprendizaje verbal y procesamiento psicomotor. Todos lo antagonistas del FNT alfa (pentoxifilina, rolipram, y otros) probados han sido inefectivos. Memantina, un antagonista no competitivo de los receptores NMDA, ha mostrado en varios estudios atenuar la capacidad del VIH para causar injuria neuronal. Un estudio preliminar sugiere que, este agente puede prevenir la declinación de la función cognitiva en sujetos con infección VIH con niveles detectables de ARN del VIH en LCR (81,82).
A pesar de los avances obtenidos en los últimos años en el tratamiento de la infección por VIH, el diagnóstico, pero sobre todo el tratamiento de DVIH, siguen siendo un reto para la comunidad científica; pues su tendencia a incrementarse en los años que vienen originará una sobrecarga de los establecimientos de salud, lo cual nos obliga a estar atentos y preparados para reconocer tempranamente la DVIH. En este sentido, es indispensable la aplicación de pruebas cognitivas breves (HDS o IHDS), las cuales deben ser conocidas por el equipo multidisciplinario de los servicios de atención al paciente con infección VIH/sida, quienes llamarán la atención y podrán hacer las referencias oportunas al médico especialista en neurología, para confirmar o descartar la sospecha de DVIH, mediante la evaluación amplia y exhaustiva de pruebas cognitivas especializadas y el manejo eficiente de cada caso en particular .
REFERENCIAS BIBLIOGRÁFICAS
1. Snider DW, Simpson DM, Nielsen S, Gold JW, Metroka CE, Posner JB. Neurological complications of acquired immune deficiency: analysis of 50 patients. Ann Neurol. 1983;14:403-18. [ Links ]
2. Simpson DM, Berger JR. Neurologic manifestations of HIV infection. Med Clin North Am. 1996;80:1363-94. [ Links ]
3. Wright D, Schneider A, Berger JR. Central nervous system opportunistic infections. Neuroimag Clin North Am. 1997;3:513-25. [ Links ]
4. Clifford DB. Neurologic complications of human immunodeficiency virus infection. The Neurologist. 1998;4:54-65. [ Links ]
5. Geraci AP, Di Roccco A, Simpsom DM. Neurologic complications of AIDS. The Neurologist. 2001;7:82-97. [ Links ]
6. Mc Arthur JC. Neurologic manifestations of AIDS. Medicine (Baltimore).1987;66:407-37. [ Links ]
7. Berger JR, Moskowitz L, Fischl M, Kelley RE. Neurologic disease as the presenting manifestation of acquired immunodeficiency syndrome. South Med J. 1987;80:683-6. [ Links ]
8. Gray F, Gherardi R, Scaravilli F. The neuropathology of the acquired immunodeficiency syndrome (AIDS): A review. Brain. 1988;111:245-66. [ Links ]
9. Lang W, Miklossy J, Deruaz JP, Pizzolato GP, Probst A, Schaffner T, et al. Neuropathology of the acquired immunodeficiency syndrome (AIDS): a report of 135 consecutive autopsy cases from Switzerland. Acta Neuropathol (Berl). 1989;77:379-90. [ Links ]
10. Lantos PL. Mc Laughlin JE, Schoitz CL, Berry CL, Tighe JR. Neuropathology of the brain in the HIV infection. Lancet. 1989;11:309-11. [ Links ]
11. Simpson DM. Human immunodeficiency virus-associated dementia: Review of pathogenesis, prophylaxis, and treatment studies of zidovudine therapy. Clin Infect Dis. 1999;29:19-34. [ Links ]
12. Report of a Working Group of the American Academy of Neurology Adquired Immune Deficiency Syndrome Task Force: Nomenclature and research case definitions for neurologic manifestations of human immunodeficiency virus type 1 (HIV-1) infection. Neurology. 1991;41:778-85. [ Links ]
13. Levy RM, Rothholtz V. HIV-1-related neurologic disorders: neurosurgical implications. Neuroimag Clin North Am. 1997;3:527-59. [ Links ]
14. Belman AL: Infants, children and adolescents. En: Berger JR, Levy RM, eds. AIDS and the Nervous System. 2 ed. Philadelphia: Lippincott-Raven; 1997. p. 223. [ Links ]
15. 16Janssen RS, Cornblath DR, Epstein LG, Foa RP, Mc Arthur JC, Price RW, et al. Human immunodeficiency virus (HIV) infection and the nervous system: report from the American Academy of Neurology AIDS Task Force (review). Neurology. 1989;39:119. [ Links ]
16. 17World Health Organization. 1990 World Health Organization consultation on the neuropsychiatric aspects of HIV-1 infection. AIDS. 1990;4;935. [ Links ]
17. 15Price RW. Neurologic disease. En: Dolin R, Masur H, Saag MS, eds. AIDS Therapy. 2 ed. New York: Churchill Livingstone; 1998. p. 622. [ Links ]
18. Dunlop O, Bjorklund R, Bruun JN, Evensen R, Goplen AK, Liestol K, et al. Early psychomotor slowing predicts the development of HIV dementia and autopsy –verified HIV encephalitis. Acta Neurol Scand. 2002;105:270-5. [ Links ]
19. Kim HJ, Kim SY, Lee K-B, Lee KW, Oh M, Choe K. Neurologic complications of human immunodeficiency virus-type 1 infection. J Korean Med Sci. 2003;18:149-57. [ Links ]
20. Mac Arthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, et al. Dementia in AIDS patients: incidence and risk factors. Neurology. 1993;43:2245-52. [ Links ]
21. Centers for Disease Control. Revision of the CDC surveillance case definition for acquired immunodeficiency syndrome. MMWR. 1987;36(suppl 1S):3S-15S. [ Links ]
22. Grant I, Heaton RK, Atkinson JH, for the HIV Neurobehavioral Research Center Group. Neurocognitive disorders in HIV-1 infection. Curr Top Microbiol Inmunol. 1995;2002:11-32. [ Links ]
23. Sacktor N, Lyles RH, Skolasky R, Kleeberger C, Selnes OA, Miller EN, et al. HIV-associated neurologic disease incidence changes: multicenter AIDS Cohort Study, 1990-1998. Neurology. 2001;56:257-60. [ Links ]
24. Jellinger KA, Setinek U, Drlicek M, Bohm G, Steurer A, Lintner F. Neuropathology and general autopsy findings in AIDS during the last 15 years. Acta Neuropathol (Berl). 2000;100:213-20. [ Links ]
25. Bouwman FH, Skolasky RL, Hes D, Selenes OA, Glass JD, Nance-Sproson TE, et al. Variable progression of HIV-associated dementia. Neurology. 1998;50:1814-20. [ Links ]
26. Bell JE, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexual with AIDS. Brain. 1998;121:2043-52. [ Links ]
27. Concha M, Selnes OA, Vlahov D. Comparison of neuropsychological performance between AIDS-free injecting drug users and homosexual men. Neuroepidemiology. 1997;16:78-85. [ Links ]
28. Marder K, Liu X, Stern Y. Risk of human immunodeficiency virus type 1-related neurologic disease in a cohort of intravenous drug users. Arch Neurol. 1995;52:1174-82. [ Links ]
29. Starace F, Baldassare C, Biancolili V. Early neuropsychological impairment in HIV-seropositive intravenous drugs users: evidence from the Italian Multicentre Neuropsychological HIV Study. Acta Psychiatr Scand. 1998;97:132-8. [ Links ]
30. Stem RA, Silva SG, Chaisson N, Evans DL. Influence of cognitive reserve on neuropsychological functioning in asymptomatic human inmunodeficiency virus-1 infection. Arch Neurol. 1996;53:148-53. [ Links ]
31. Maj M, Satz P, Janssen R. WHO Neuropsychiatric AIDS Study, cross-sectional phase II: neuropsychological and neurological findings. Arch Gen Psychiatry. 1994;51:51-61. [ Links ]
32. Satz P, Morgenstem H, Miller EN. Low education as possible risk factor for cognitive abnormalities in HIV-1: findings from the Multicenter AIDS Cohort Study (MACS). J Adquir Immune Defic Syndr. 1993;6:503-11. [ Links ]
33. De Ronchi D, Faranca I, Berardi D. Risk factors for cognitive impairment in HIV-1-Infected persons with different risk behaviors. Arch Neurol. 2002;59:812-8. [ Links ]
34. Stern Y, McDermott MP, Albert S. Factors associated with incident human immunodeficiency virus dementia. Arch Neurol. 2001;58:473-9. [ Links ]
35. Beach RS, Morgan R, Wilkie F. Plasma vitamin B12 level as a potential cofactor in studies of human immunodeficiency virus Type1-related cognitive changes. Arch Neurol. 1992;49:501-6. [ Links ]
36. Marder K, Stern Y, Malouf R. Neurologic and neuropsychological manifestations of human immunodeficiency virus infection in intravenous drug users without acquired immunodeficiency syndrome: relationship to head injury. Arch Neurol. 1992;49:1169-75. [ Links ]
37. Valcour V, Shikuma C, Shiramizu B, Waters M, Poff P, Selnes OA, et al. Higher frequency of dementia in older HIV-1 individuals: The Hawaii Aging with HIV-1 cohort. Neurology. 2004;63:822-7. [ Links ]
38. Valcour V, Shikuma C, Shiramizu B, Waters M, Poff P, Selnes OA, et al. Age, Apolipoprotein E4 and the risk of HIV Dementia: The Hawaii Aging with HIV-1 cohort. J Neuroinmunology. 2004;157:197-202. [ Links ]
39. Navia BA, Jordan BD, Price RW. The acquired immune deficiency syndrome dementia complex: Clinical features. Ann Neurol. 1986;19:517-24. [ Links ]
40. McArthur JC. HIV Dementia: an evolving disease. J Neuroinmunology. 2004;157:3-10. [ Links ]
41. Power C, Selnes OA, Grim JA, McArthur JC. The HIV Dementia Scale: a rapid screning test. J AIDS. 1995;8:273-8. [ Links ]
42. Sacktor NC, Wong M, Nakasujja N, Skolasky RL, Selnes OA, Musisi S, et al. The International HIV Dementia Scale: a new rapid screening test for HIV dementia. AIDS. 2005;19:1367-74. [ Links ]
43. Sacktor N, Schifitto G, Mc Dermott MP, Marder K, Mc Arthur JC. Kieburtz K. Transdermal selegiline in HIV-asssociated cognitive impairment: pilot, placebo controlled study. Neurology. 2000;54:233-5. [ Links ]
44. Marder K, Albert SM, Mc Dermoth MP, Mc Arhur JC, Schifitto G, Selnes OA, et al. Inter-rater reliability of a clinical staging of HIV-associated cognitive impairment. Neurology. 2003;60:1467-73. [ Links ]
45. The Dana Consortium on Therapy for HIV Dementia and Related Cognitive Disorders. Clinical confirmation of the American Academy of Neurology algorithm for HIV-1-associated cognitive/motor disorder. Neurology. 1996;47:1247-53. [ Links ]
46. Fahn S, Marsden S, Calne D. Recent Developments in Parkinsons disease. Florham Park: Macmillan Healthcare Information; 1987. [ Links ]
47. Rey A. Lexamen psychologique dans les cas dencephalopathie traumatique. Arch Psychologie. 1941;28:286-340. [ Links ]
48. Wechsler D. Wechsler Adult intelligence Scale-Revised. New York: The Psychological Corporation; 1981. [ Links ]
49. Miller EN, Satz P. VisScher B. Computerized and conventional neuropsychological assesment of HIV-1 infected homosexual men. Neurology. 1991;41:1608-16. [ Links ]
50. Klove H. Clinical neuropsychogy. Med Clin North Am. 1963;46:1647-58. [ Links ]
51. Benton AL, Hamsher KD. Multilingual Aphasia Examination. Iowa City: University of Iowa; 1976.
52. Richards M, Cote LJ, Stern Y. Executive function in Parkinsons diasease: seth-shifting or set-maintenance? J Clin Exp Neuropsychol. 1993;15:266-79.
53. Lawton MP, Brody EM. Assesment of older people: self-maintaining and instrumental activities of daily living. Gerontologist. 1969;9:179-86.
54. Katz S, Ford A, Moskowitz R. Studies of illness in the aged: the index of ADL. JAMA. 1963;183:914-9.
55. Beck AT, Steer R, Brown J. BDI-II Manual. San Antonio. The Psychological Corp. 1996.
56. Portegies P, Eipstein LG, Hunt STA, de Gans J, Goudsmith J. Human immunodeficiency virus type 1 antigen in cerebrospinal fluid: correlation with clinical neurologic status. Arch Neurol. 1989;46:261-4.
57. Del Pan GJ, Mc Arthur JH, Aylward E, Selnes OA, Nance-Sproson TE, Kumar AJ, et al. Pattterns of cerebral atrophy in HIV-1-infected individuals: results of a quantitative MRI analisis. Neurology. 1992;42:2125-30.
58. Patel SH, Kolson DL, Glosser G, Matozzo I, Ge Y, Bab JS, et al. Correlation between percentage of brain parenchymal volume and neurocognitive performance in HIV-Infected patients. Am J Neuroradiol. 2002;23:543-9
59. Yiannoutsos CT, Ernst T, Chang L, Lee PL, Richards T, Marra CM, et al. Regional patterns of brain metabolites in AIDS dementia complex. Neuroimage. 2004;23:928-35.
60. Avison MJ, Nath A, Berger JR. Understanding pathogenesis and treatment of HIV dementia: a role for magnetic resonance? TNS. 2002;25:468-73.
61. Turner BG, Summers MF. Structural Biology of HIV. J Mol Biol. 1999;285:1-32.
62. Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Research. 2005;111:194–213.
63. González-Scarano F, Martín-García J. The neuropathogenesis of AIDS. Nature Reviews Immunology. 2005;5:69-81.
64. Albright AV, Soldan SS, Gonzales-Scarano F. Pathogenesis of human immunodeficiency virus-induced neurological disease. J Neurobiology. 2003;9:222-7.
65. Lavi1 E, Kolson DL, Ulrich1 AM, Fu L, Gonzalez-Scarano F. Chemokine receptors in the human brain and their relationship to HIV infection. Journal of NeuroVirology. 1998;4:301-11.
66. Brandimarti R, Khan MZ, Fatatis A, Meucci O. Regulation of cell cycle proteins by chemokine receptors: A novel pathway in human immunodeficiency virus neuropathogenesis? J NeuroVirology. 2004;10(suppl.1):1–5.
67. Toborek M, Lee YW, Flora G, Pu H, András IE, Wylegala E, et al. Mechanisms of the blood–brain barrier disruption in HIV-1 infection. Cellular and Molecular Neurobiology. 2005;25:288-98.
68. Dieter J. Meyerhoff. Effects of alcohol and HIV infection on the central nervous system. Alcohol Research & Health. 2001;25:181-99.
69. Fotheringhama J, Maynea M, Holdena C, Nathd A, Geigerae JD. Adenosine receptors control HIV-1 Tat-induced inflammatory responses through protein phosphatase. Virology. 2004;327:186–95.
70. Van Marle G, Henry S, Todoruka T, Sullivan A, Silva C, Rourkec SB, et al. Human immunodeficiency virus type 1 Nef protein mediates neural cell death: a neurotoxic role for IP-10. Virology. 2004;329:302–18.
71. Mordelet E, Kissa K, Cressant A, Gray F, Ozden S, Vidal C, et al. Histopathological and cognitive defects induced by Nef in the brain. The FASEB J. 2004;18:1851-61.
72. Price TO, Ercal N, Nakaoke R, Banks WA. HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Research. 2005;104:57-63.
73. Turchan J, Pocernich CB , Gairola C, Chauhan A, Schifitto G, Butterfield DA, et al. Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology. 2003;60:307–14.
74. Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, DEmilia DM, et al. Caspase Cascades in Human Immunodeficiency Virus-Associated Neurodegeneration. The Journal of Neuroscience. 2002;22(10):4015–24.
75. Sacktor N, Tarwater PM, Skolasky RL, McArthur JC, Selnes OA, Becker J, et al. CSF antiretroviral drug penetrance and the treatment of HIV-associated psychomotor slowing. Neurology. 2001;57:542-4.
76. Brew BJ, Halman M, Catalán J, Sacktor N, Price RW, Brown S, et al. Abacavir in AIDS Dementia Complex: Efficacy and lessons for future trials. Neurology (in press).
77. Cohen RA, Boland R, Paul R, Tashima KT, Schoenbaum EE, Celentano DD, et al. Neurocognitive performance enhanced by highly active antiretroviral therapy in HIV-infected women. AIDS. 2001;15:341-5.
78. Tozzi V, Balestra P, Galgani S, Narciso P, Ferri F, Sebastián G, et al. Positive and sustained effects of highly active antiretroviral therapy on HIV-associated neurocognitive impairment. AIDS. 1999;13:1889-97.
79. Sacktor N, McDermott MP, Marder K, Schifitto G, Selnes OA, McArthur JC, et al. HIV-associated cognitive impairment before and after the advent of combination therapy. J Neurovirology. 2002;8:136–42.
80. Navia BA, Dafni U, Simpson D, Tucker T, Singer E, McArthur JC, et al. A phase I/II trial with nimodipine for HIV-related neurological complications. Neurology. 1998;19:517-24.
81. Lipton SA. Memantina prevents HIV coat protein-induced neuronal injury in vitro. Neurology. 1992; 42:1403-5.
82. Nath A, Haughey NJ, Jones M, Anderson C, Bell JE, Geiger JD. Synergistic neurotoxicity by human inmunodeficiency virus proteins Tat and gp120: Protection by memantine. Ann Neurol. 2000;47:186-94.
Manuscrito recibido el 09 de agosto de 2006 y aceptado para publicación el 01 setiembre de 2006.
Correspondencia:
Nilton Custodio Capuñay
Correo-e: niltoncustodio@neuroconsultas.com

