










Serviços Personalizados
Journal

Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkAnales de la Facultad de Medicina
versão impressa ISSN 1025-5583
An. Fac. med. vol.80 no.3 Lima jul./set. 2019
http://dx.doi.org/10.15381/anales.803.16861
REPORTE DE CASOS
Rabdomiosarcoma embrionario parameningeo en adulto
Parameningeal embryonic rhabdomyosarcoma in adult
Jose Huamán Muñante1,2,a, Rolig Aliaga Chávez2,b, Julio Grados Doroteo2,b, Gonzalo Huamán Junco3,c
1 Facultad de Medicina, Universidad Nacional Mayor de San Marcos. Lima, Perú.
2 Hospital Nacional Arzobispo Loayza. Lima, Perú.
3 Facultad de Medicina, Universidad Ricardo Palma. Lima, Perú.
a Médico internista, ORCID: 0000-0002-8712-0376.
b Médico oncólogo
c Estudiante de medicina, ORCID: 0000-0001-7604-7831.
RESUMEN
Paciente mujer de 22 años, con tiempo de enfermedad de 2 meses, presentó epífora, eritema conjuntival leve, prurito en ojo derecho y aparición de pequeña tumoración verdosa a nivel del canto interno asociada a edema bipalpebral leve. El edema progresó limitando la visión en dicho ojo, asociándose la protrusión del globo ocular derecho. Se realizó estudio anatomopatologico del tumor nasosinusal presentando nidos celulares hipercromáticos de aspecto infiltrativo y el estudio inmunohistoquímico concluyó como rabdomiosarcoma embrionario. Se realizó quimioterapia.
Palabras clave: Rabdomiosarcoma Embrionario; Adulto; Diagnóstico (fuente: DeCS BIREME).
ABSTRACT
A 22-year-old female patient, with 2-month disease time, presented with epiphora, mild conjunctival erythema and pruritus in the right eye. A small greenish-colored tumor appeared at the level of the internal canthus associated with mild bipalpebral edema. The edema progresses limiting the vision in said eye, associating the protrusion of the right eyeball. An anatomopathological study of the nasosinusal tumor was performed, presenting hyperchromatic cellular nests with an infiltrative aspect and the immunohistochemical study concluded as embryonic rhabdomyosarcoma. Chemotherapy was performed.
Keywords: Rhabdomyosarcoma, Embryonal; Adult; Diagnosis (source: MeSH NLM).
INTRODUCCIÓN
El rabdomiosarcoma (RMS) es uno de los sarcomas de tejidos blandos más comunes (STB) en la población pediátrica, que representa más de 50% de todas los STB en este grupo de edad, con una incidencia de 0,4414 por 100 000 por año en los Estados Unidos. Los sarcomas en Perú constituyen aproximadamente entre 0,7 a 1% del total de tumores malignos y son responsables del 2% de la mortalidad debido al cáncer. El RMS en adultos, sin embargo, ocurre con menos frecuencia, ya que comprende solo el 3% de los STB y menos del 1% de todas las neoplasias malignas (1). Se estima 350 casos nuevos por año de RMS en los Estados Unidos; la mitad de estos son diagnosticados en pacientes menores de 10 años. El RMS puede surgir en una variedad de sitios anatómicos, incluyendo la región de cabeza y cuello (35-40%), la genitourinaria (25%), tronco y extremidades (20%) (2). La tasa de supervivencia a 5 años con tratamiento en adultos (27%) continúa muy por debajo respecto a los niños (70%) (1).
Presentamos un caso infrecuente de una paciente mujer adulta joven con el diagnóstico de rabdomiosarcoma embrionario primario parameníngeo.
REPORTE DE CASO
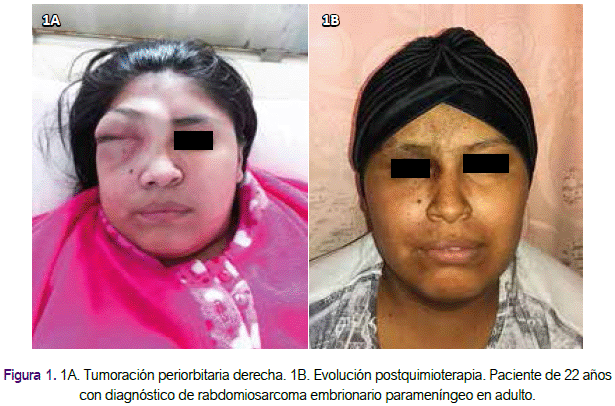
Paciente mujer de 22 años, nacida y procedente de Piura (norte del Perú), sin antecedentes de importancia, con un tiempo de enfermedad de 2 meses, insidiosa y progresiva caracterizada por presentar epífora, eritema conjuntival leve y prurito en ojo derecho, seguido de la aparición de una pequeña tumoración a nivel del canto interno asociada a edema bipalpebral leve. El edema progresa limitando la visión en dicho ojo, asociándose protrusión del globo ocular derecho (Figura 1A).
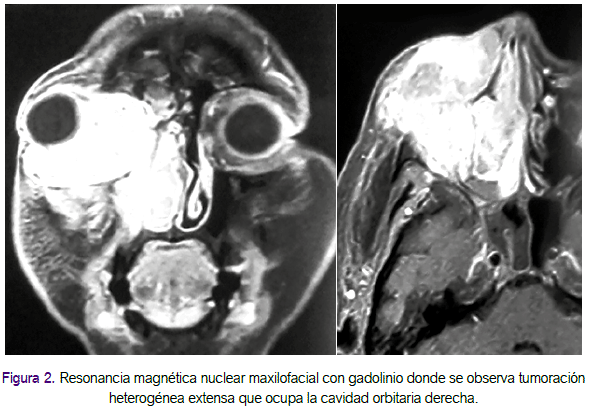
Los resultados de los exámenes auxiliares, al inicio el hemograma sin alteraciones y 2 semanas después leucocitos en 17 620/mL, abastonados 3% y segmentados 87%. La ecografía de partes blandas periorbitaria concluyó: tumoración periorbitaria derecha abscedada. En la resonancia magnética maxilofacial con gadolinio se observó tumoración heterogénea extensa que ocupaba la cavidad orbitaria derecha condicionando proptosis anterior y lateral con extensión hacia senos etmoidales, cavidad nasal y parcialmente al seno maxilar del mismo lado aproximadamente de 6 cm (Figura 2).
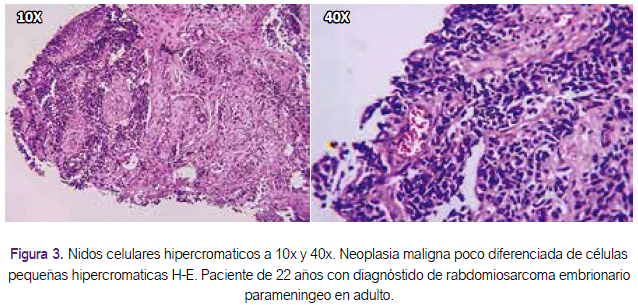
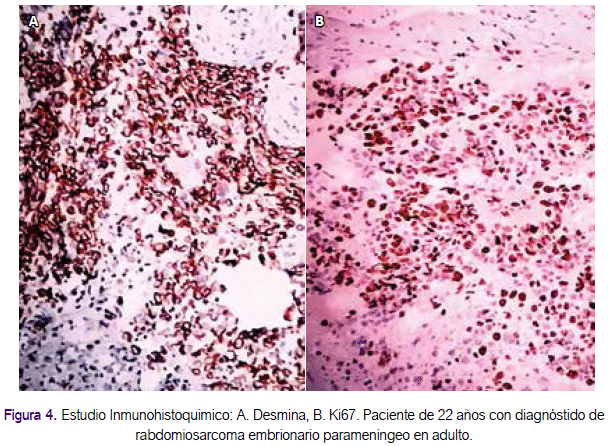
Se realizó estudio anatomopatológico de la muestra del tumor nasosinusal que presentó nidos celulares hipercromáticos de aspecto infiltrativo, determinando una neoplasia maligna poco diferenciada de células pequeñas hipercromáticas (Figura 3). El estudio inmunohistoquímico reveló los siguientes hallazgos: desmina positivo, myoD1 positivo, mioglobina positiva, CD20 negativo, CD3 negativo, Ki67 70%, concluyendo: rabdomiosarcoma embrionario (Figura 4). El resto de estudios de imágenes tomográficas de tórax y abdomen, para estadiaje, fueron negativos. No se realizó estudio de punción lumbar. El estadio para este caso fue: IIB (T2a o T2b, N0, M0, G3).
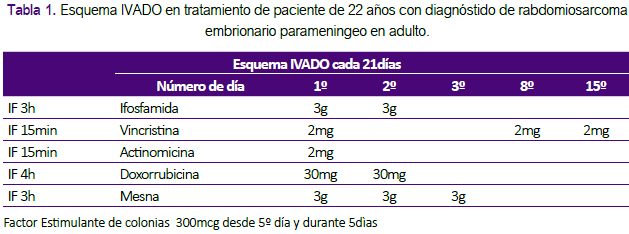
Se indicó tratamiento antibiótico con vancomicina y meropenem por 2 semanas por el absceso; y se inició quimioterapia con el esquema IVADO (ifosfamida 3 g/m2 día 1-2, mesna 3 g/m2 día 1-2, vincristina 1,5 mg/m2 día 1, actinomicina 1,5 mg/m2 día 1 y doxorrubicina 30mg/m2 día 1-2 ) cada 21 días (Tabla 1). Luego de 4 cursos presentó respuesta clínica y por imágenes completa bajo los criterios de evaluación radiológica con los criterios de RECIST (Figura 1B). Presentó neutropenia grado 2, emesis grado 1 y alopecia asociadas al tratamiento. La paciente fue referida a otra institución de mayor resolución para completar tratamiento de consolidación con radioterapia o la opción de resección quirúrgica.
DISCUSIÓN
El RMS es un tumor de tejido blando altamente maligno que se pensó que derivaba del músculo esquelético; sin embargo, ahora se sabe que se originan de los rabdomioblastos tempranos que se pueden encontrar en una amplia variedad de lugares en todo el cuerpo con un grado variable de diferenciación (1,3).
Los síntomas clínicos varían y dependen de la ubicación del tumor primario y de la presencia o ausencia de metástasis. El RMS de cabeza y cuello puede presentarse de forma indolente, con signos y síntomas inespecíficos o mínimos, lo que lleva a un retraso en el diagnóstico. Por ejemplo, la otitis crónica u obstrucción nasal puede ser la presentación de una localización nasofaríngea, mientras que la cefalea persistente pueden ser un síntoma de extensión del tumor intracraneal. Existen tres grupos de RMS, según la histología: embrionario, alveolar y pleomórfico, de los cuales los dos primeros se producen principalmente en niños y el último sólo en adultos (4). El RMS embrionario (RMSe) representa aproximadamente el 70% de los casos, afecta a pacientes más jóvenes y tiene un mejor pronóstico. El RMS alveolar (RMSa) representa aproximadamente el 15%, se observa en niños mayores y tiene un peor pronóstico. Se reconocen tres ubicaciones primarias diferentes de RMS en cabeza y cuello: parameníngeo (PM), no parameníngeo (NPM) y orbital (ORB), que están involucradas en aproximadamente el 50%, 25% y 25% de los casos, respectivamente. La ubicación PM conlleva un peor pronóstico (supervivencia general a 5 años 64%–73%) en comparación con las ubicaciones NPM y ORB (supervivencia general de 5 años 64%-80% y 85–88%, respectivamente) por al menos dos razones. En primer lugar, la ubicación profunda puede ocultar un tumor que permanece clínicamente silencioso a menos que haya crecido a un tamaño considerable y sea visible desde el exterior, y en segundo lugar porque la ubicación del PM está muy cerca de la base del cráneo y contiene estructuras vitales como la arteria carótida interna, vena yugular interna y múltiples nervios craneales, lo que excluye la resección oncológica. Las ubicaciones PM están en contacto directo o cercano con la base del cráneo, dichos tumores pueden originarse en la nasofaringe, el espacio parafaríngeo, la fosa infratemporal, la cavidad nasal, los senos paranasales, el mastoideo o el oído medio. El RMSe PM puede mostrar una propagación más temprana a la base del cráneo y al compartimiento intracraneal en comparación con otros lugares, y una evaluación precisa de la extensión local es uno de los principales objetivos de la imagen. La supervivencia es peor y las opciones de tratamiento difieren cuando hay afectación de la base del cráneo o extensión del tumor intracraneal (5).
Los tumores de la cabeza y el cuello se representan mejor con resonancia magnética nuclear (RMN). Posee un contraste superior de los tejidos blandos, excelente resolución espacial, imágenes multiplanares y la ausencia de radiación ionizante de efectos potencialmente dañinos, hacen de la RMN un instrumento fundamental para el estadiaje local preciso (5).
Las características histopatológicas del RMSe se presentan como foco celular denso compuesto de células poligonales y fusiformes con contornos nucleares irregulares (RMSe denso) mientras RMSa está compuesto por células monomorfas con contornos redondos, incluso nucleares (RMSa sólidas) agrupadas en nidos separados por septos fibrosos (6).
Los estudios inmunohistoquímicos constituyen el principal examen diagnóstico. Los marcadores que se encuentran típicamente en RMS incluyen anticuerpos contra desmina (90%), actina específica del músculo, myoD1 (71%–91%) y mioglobina. La miogenina (90%) se expresa más en el alveolar que en el embrionario. Los tipos de vimentina y desmina son generalmente positivos pero menos específicos, ya que pueden ser positivos en otros tumores con diferenciación del músculo esquelético. Miogenina y MyoD1 son reguladores de la transcripción miogénicos que se expresan temprano en la diferenciación del músculo esquelético (antes que la desmina, la actina, la mioglobina y la miosina) y son sensibles y específicos para los rabdomiosarcomas (4,7,8,9).
No hay un régimen de quimioterapia estandarizado para pacientes adultos con RMS. Los estudios informan que los adultos pueden obtener buenos resultados con los regímenes pediátricos, especialmente para adultos que se presentan con RMSe o RMSa. Poco se sabe sobre la quimioterapia óptima para pacientes con RMS pleomórficos. En la población adulta, se han usado otras combinaciones de quimioterapia además de vincristina, actinomicina D y ciclofosfamida habitualmente utilizada en la población pediátrica; estos diferentes esquemas se han reportado en pequeñas series de población y no han tenido mayor impacto en la supervivencia global. La combinación de medicamentos utilizados en aquellos estudios incluye ciclofosfamida, doxorubicina y vincristina; además ifosfamida más doxorubicina; o doxorubicina, ifosfamida y dacarbazina (2,10).
En el estudio analítico retrospectivo de Fischer y col. en EEUU, se investigó la terapia multimodal (resección quirurgica + quimioterapia + radiación) a nivel nacional. Se incluyó 2 312 pacientes de los cuales el 44% fueron pediátricos (edad <15 años), 22% adolescentes y adultos jóvenes (edades 15–39), y 34% adultos (edad ≥ 40 años). Esta terapia se asoció de forma independiente con mejor supervivencia a los 5 años tanto para adolescentes y adultos jóvenes como para adultos mayores con RMS, similar a los pacientes pediátricos, pero la terapia multimodal es infrautilizada (1). En nuestro caso, la paciente completó 4 cursos de poliquimioterapia IVADO con respuesta clínica y por imágenes completa, por lo que se refirió a otra institución de mayor resolución para completar el tratamiento de consolidación con radioterapia ver sus cirugía y a posterioridad discutir la posibilidad de tratamiento adyuvante y/o mantenimiento sabiendo que estos tumores en edad adulta son altamente recurrentes.
REFERENCIAS BIBLIOGRÁFICAS
1. Fischer TD, Gaitonde SG, Bandera BC, Raval MV, Vasudevan SA, Gow KW, et al. Pediatric-protocol of multimodal therapy is associated with improved survival in AYAs and adults with rhabdomyosarcoma. Surgery. 2018;163(2):324-9. DOI: 10.1016/j.surg.2017.10.027. [ Links ]
2. Ruiz-Mesa C, Goldberg JM, Coronado Munoz AJ, Dumont SN, Trent JC. Rhabdomyosarcoma in Adults: New Perspectives on Therapy. Curr Treat Options in Oncol. 2015;16(6):27. DOI: 10.1007/s11864-015-0342-8. [ Links ]
3. Häußler SM, Stromberger C, Olze H, Seifert G, Knopke S, Böttcher A. Head and neck rhabdomyosarcoma in children: a 20-year retrospective study at a tertiary referral center. J Cancer Res Clin Oncol. 2018;144(2):371-9. DOI: 10.1007/s00432-017-2544-x. [ Links ]
4. Paulino AC, Okcu MF. Rhabdomyosarcoma. Curr Probl Cancer. 2008;32(1):7-34. DOI: 10.1016/j.currproblcancer.2007.11.001. [ Links ]
5. Freling NJM, Merks JHM, Saeed P, Balm AJM, Bras J, Pieters BR, etal. Imaging findings in craniofacial childhood rhabdomyosarcoma. Pediatr Radiol. 2010;40(11):1723- 38. DOI: 10.1007/s00247-010-1787-3. [ Links ]
6. Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013;20(6):387-97. DOI: 10.1097/PAP.0b013e3182a92d0d. [ Links ]
7. Jurdy L, Merks JHM, Pieters BR, Mourits MP, Kloos RJHM, Strackee SD, et al. Orbital rhabdomyosarcomas: A review. Saudi Journal of Ophthalmology. 2013;27(3):167-75. DOI: 10.1016/j.sjopt.2013.06.004. [ Links ]
8. Córdoba Rovira SM, Inarejos Clemente EJ. Rabdomiosarcoma infantil. Radiología. 2016;58(6):481-90. [ Links ]
9. Alberto Mejía-Salas J, Sánchez-Corona H, PriegoNiño A, Cárdenas-Rodríguez E, Antonio SánchezGalindo J. Primary testicular rhabdomyosarcoma: A case report. Cirugía y Cirujanos. 2017;85(2): 143-147. DOI: 10.1016/j.circen.2017.02.002. [ Links ]
10. Ge X, Huang D-S, Shi J-T, Ma J-M. Multidisciplinary Collaborative Therapy for 30 Children with Orbital Rhabdomyosarcoma. Asian Pacific Journal of Cancer Prevention. 2013;14(8):4641-6. DOI: 10.7314/apjcp.2013.14.8.4641. [ Links ]
Conflictos de interés: Los autores declaran no tener conflictos de interés.
Fuente de financiamiento: Autofinanciado
Correspondencia:
José Gonzalo Huamán Muñante
josgon975@hotmail.com
Recibido: 14 de mayo 2019
Aceptado: 23 de junio 2019
Publicación en línea: 30 de setiembre 2019

