











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
La histiocitosis sinusal con linfadenopatía masiva (HSLM), también conocida como enfermedad de Rosai-Dorfman (ERD), fue descrita por primera vez por Rosai y Dorfman en el año 1969 1. Se trata de una enfermedad proliferativa histiocítica benigna poco común y de etiología desconocida que se caracteriza por la presencia de linfadenopatía cervical periférica bilateral, indolora, masiva 2. Los pacientes con ERD también pueden presentar otros síntomas como fiebre y alteraciones laboratoriales como neutrofilia, aumento de la velocidad de sedimentación de eritrocitos séricos (VSG), leucocitosis, linfopenia, hiperglobulinemia policlonal y anemia 3. La presentación extraganglionar, que se observa en una minoría de casos, puede afectar prácticamente a cualquier sitio anatómico, pero suele asociarse con linfadenopatía concomitante 3,4. La ERD ósea en ausencia de linfadenopatía, es decir, enfermedad primaria ósea, representa menos del 1% de todos los casos 3 y se presenta como una lesión solitaria en la mayoría de los pacientes 5. El diagnóstico definitivo se basa en el estudio histológico de las lesiones para identificar la presencia de histiocitos grandes, característicos, dentro de un infiltrado celular inflamatorio crónico denso 6.
Se presenta un caso de enfermedad de Rosai-Dorfman primaria ósea y se describen los principales hallazgos radiológicos e histopatológicos encontrados, así como el tratamiento recibido.
REPORTE DE CASO
Paciente mujer de 38 años sin antecedentes de importancia con historia de 3 meses de dolor en miembro inferior derecho, de mayor intensidad por las noches y al realizar esfuerzo, que calmaba inicialmente con el uso de antiinflamatorios no esteroideos (AINES). La intensidad del dolor fue incrementándose progresivamente hasta llegar a presentar dificultad para la marcha, por lo que decidió acudir al servicio de emergencia. Al examen físico se evidenció dolor a la digitopresión en la cara anterior del tercio distal del muslo así como limitación a la movilización activa y pasiva del miembro inferior derecho. No presentaba adenopatías poplíteas, inguinales, axilares ni cervicales.
Como estudio inicial se tomaron radiografías simples de rodilla en proyección frontal y lateral, hallándose una lesión ósea lítica en la metáfisis y epífisis distal del fémur derecho (Figura 1).
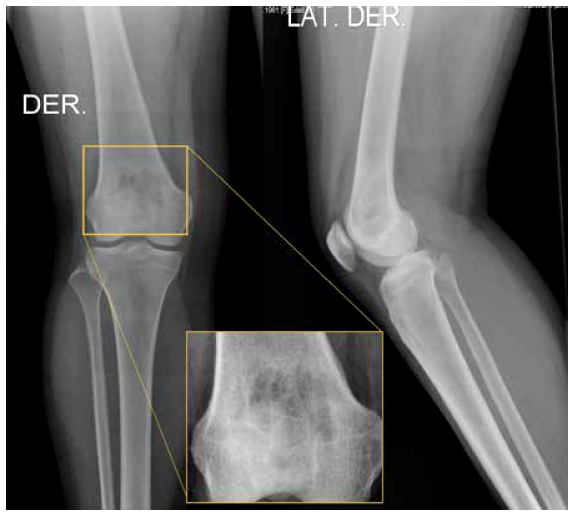
Figura 1 Radiografía simple de rodilla derecha, vistas frontal y lateral. Se evidencia una lesión ósea lítica parcialmente circunscrita, de bordes poco definidos, localizada en la metáfisis y epífisis distal del fémur derecho.
En relación al hallazgo descrito, se decidió complementar el estudio mediante tomografía computarizada (TC) de miembros inferiores. Se observó una lesión osteolítica a nivel de la metáfisis y epífisis distal del fémur derecho, de localización central, de 47x45x47 mm; no se identificó presencia de matriz ósea ni condral, presentaba algunos septos internos y mostraba bordes irregulares parcialmente escleróticos. Además, condicionaba disrupción de la cortical ósea e impresionaba comprometer las partes blandas adyacentes (Figuras 2 y 3).
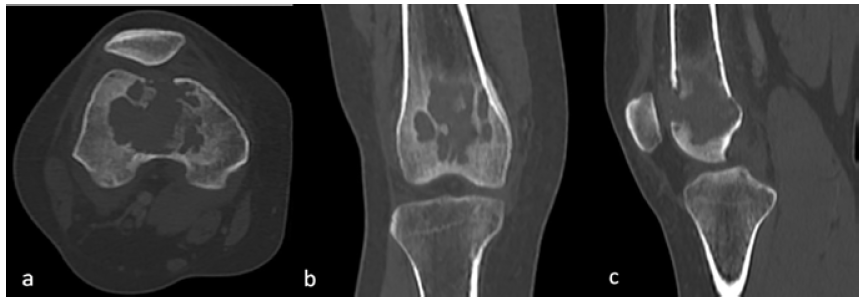
Figura 2 TC de miembros inferiores, ventana ósea. Las vistas axial (a), coronal (b) y sagital (c) muestran la presencia de una lesión lítica de márgenes irregulares a nivel de la metáfisis y epífisis distal del fémur derecho.
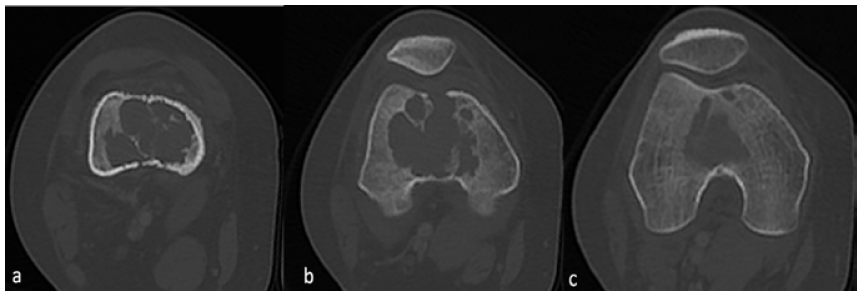
Figura 3 TC de miembros inferiores, ventana ósea. Los cortes en vista axial progresivos (a,b,c) muestran que la lesión lítica tiene localización central, presenta septos internos y bordes parcialmente escleróticos; condiciona disrupción de la cortical ósea e impresiona compromiso de las partes blandas adyacentes.
Se realizó biopsia de la lesión con aguja fina mediante guía fluoroscópica. El estudio histológico mostró la presencia de tejido óseo con infiltrado inflamatorio crónico linfoplasmocítico asociado a acúmulos de histiocitos largos con emperipolesis (Figura 4), el cual fue complementado con estudio de inmunohistoquímica: proteína S100 positivo, CD1a negativo (Figura 5). Estos hallazgos fueron compatibles con la enfermedad de Rosai-Dorfman con compromiso óseo.
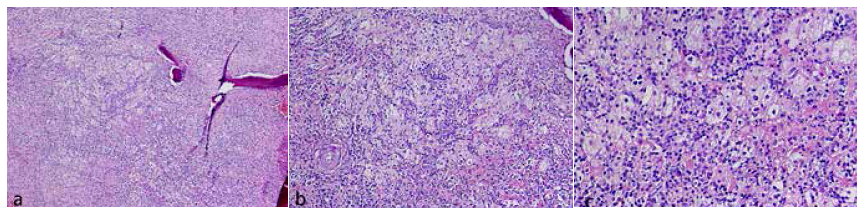
Figura 4 Estudio anatomopatológico de biopsia de la lesión descrita, cortes histológicos con tinción hematoxilina-eosina. El primero (a), a menor aumento (10x), muestra el tejido óseo comprometido. Los cortes a mayor aumento (b) (40x) y (c) (100x) muestran la presencia de histiocitos largos con emperipolesis en medio de un marcado infiltrado inflamatorio crónico, compatible con Enfermedad de Rosai-Dorfman.
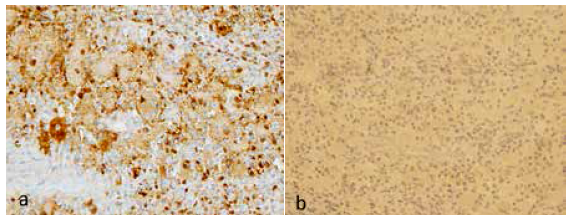
Figura 5 La imagen de la izquierda (100x) evidencia histiocitos grandes fuertemente teñidos por inmunotinción S100 que muestran claramente linfocitos intracitoplasmáticos (emperipolesis), que es característico de la enfermedad de Rosai-Dorfman. S100 delinea negativamente los linfocitos dentro del citoplasma. A la derecha (40x), CD1a negativo apoya el diagnóstico definitivo.
La paciente fue tratada mediante curetaje y electrofulguración de la lesión, así como reducción abierta y fijación interna (RAFI) con colocación de placa de compresión de bloqueo (LCP) y cementación. Tras el procedimiento la paciente fue dada de alta con seguimiento por consulta externa, en los cuales no se ha evidenciado signos de recurrencia ni otras manifestaciones de la enfermedad tras un seguimiento de 10 meses.
DISCUSIÓN
La enfermedad de Rosai-Dorfman es una entidad poco frecuente. La serie de casos más grande fue publicada por Foucar y col. en el año 1990 con 423 casos; en ella, 43% de los pacientes presentaron manifestaciones extra ganglionares, 7% de ellos con compromiso óseo y adenopatías 3. Sin embargo, la presentación ósea primaria de esta enfermedad, aislada y sin adenopatías asociadas, es incluso aún más rara (0,7%), siendo muy pocos los casos descritos en la literatura mundial 5,6,7. Estos trabajos reportan una edad media de presentación de entre 28 y 41 años 5,6, rango de edad en la que se encontraba nuestra paciente. Describen también que se presenta como una lesión ósea uni o multifocal 3,7 en distintos huesos del cuerpo, siendo la tibia, el fémur y el húmero los huesos largos comprometidos con mayor frecuencia, a nivel de su metáfisis 5,6; semejante a la lesión femoral de nuestra paciente.
Parekh en el 2016 reportó un caso de ERD primaria de hueso de lesión única en radio distal de una paciente adulta joven 8, a pesar de ser una enfermedad benigna, el comportamiento en ese caso tenía características agresivas por la destrucción cortical significativa que presentaba; en contraste Ross 9 en el 2019 reportó dos pacientes con ERD antagónicos: adulta mayor con patología poliostótica y adulto joven con patología monostótica, ambos casos no presentaban signos de agresividad, por lo que es importante considerar la heterogeneidad de las características radiológicas y de grupo etario al momento de afrontar entidades de similares características.
El estudio imagenológico puede realizarse inicialmente mediante la radiografía simple y complementarse con estudio tomográfico para definir mejor las características de la lesión. Los casos reportados describen la presencia de una lesión lítica central que involucra el canal medular, multilobulada, en algunos casos con septos internos y con un borde esclerótico variable, siendo inusual que muestre características agresivas como destrucción cortical, reacción perióstica o compromiso de partes blandas 6. El caso de nuestra paciente mostraba una lesión única, lítica, con septos internos y bordes parcialmente escleróticos; sin embargo, condicionaba disrupción de la cortical ósea y compromiso de partes blandas, por lo que contaba con algunas características de agresividad.
La inespecificidad de los hallazgos radiológicos en estos casos hace que en pacientes jóvenes se incluya en el diagnóstico diferencial la posibilidad de osteomielitis, histiocitosis de células de Langerhans (HCL), linfoma, plasmocitoma y lesiones metastásicas, por lo que resulta importante considerar los datos clínicos y laboratoriales 6. Es por ello que el diagnóstico final de esta entidad se realiza mediante el estudio histológico, siendo el hallazgo más característico la presencia de histiocitos grandes abundantes o dispersos con emperipolesis (capacidad de incluir a otras células en su citoplasma), dentro de un infiltrado crónico de células inflamatorias 3. Cuando estos histiocitos se encuentran dispersos, pueden no aparecer en la muestra de la biopsia y catalogarse como osteomielitis, requiriendo la toma de una segunda muestra 6. La inmunohistoquímica ayuda a diferenciar respecto a otras enfermedades como la histiocitosis de células de Langerhans, ya que la ERD es positiva para los marcadores CD68 y proteína S100, y negativa para el marcador CD1a.
El diagnóstico reciente de ERD ósea amerita estudios complementarios para descartar la presencia de otros focos de enfermedad, lo que incluye un examen físico completo, TC de tórax, abdomen y pelvis, así como gammagrafía ósea. En los últimos años se ha propuesto el uso de la tomografía por emisión de positrones (PET-CT) como método más sensible para la detección de lesiones no palpables con actividad metabólica activa 10.
El tratamiento estándar en los pacientes con lesiones óseas sintomáticas es el curetaje, sólo o con cementación, el cual ha demostrado controlar la extensión de la lesión y disminuir la intensidad del dolor 5. Este es el tratamiento que recibió nuestra paciente, una vez realizado el diagnóstico. Otros tratamientos no quirúrgicos descritos son la radioterapia, quimioterapia y corticoterapia a largo plazo; sin embargo, son considerados de segunda línea 11,12 por lo que no fueron utilizados en nuestra paciente.
El pronóstico de esta enfermedad en su presentación habitual es considerado benigno y los pacientes suelen tener una buena evolución. Sin embargo, la información disponible sobre los pacientes con ERD primaria ósea es mucho más limitada, siendo aún incierto si presentan una evolución similar favorable o si el riesgo de recurrencia o extensión de la enfermedad es mayor 6. En el caso de nuestra paciente, no se han registrado signos de recurrencia o extensión de la enfermedad tras 10 meses de seguimiento.
La enfermedad de Rosai-Dorfman primaria ósea es una entidad muy rara cuyo diagnóstico se apoya en el estudio por imágenes y se confirma mediante el análisis histopatológico, siendo el tratamiento principal el curetaje de la lesión. Se sugiere el desarrollo de estudios longitudinales que ayuden a esclarecer el pronóstico de estos pacientes a largo plazo.

