











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La displasia mesenquimal placentaria (DMP) es una patología obstétrica benigna poco frecuente que se caracteriza por la presencia de feto y una placenta grande para la edad gestacional por encima del 90° percentil, que ecográfica y macroscópicamente recuerda a una mola parcial 1; la superficie materna presenta vesículas de diversos tamaños a manera de “racimo de uvas” difusamente dispersas entre el parénquima, la superficie fetal contiene vasos aneurismáticos o varicosos muy dilatados. La microscopía revela degeneración hidrópica en las vellosidades troncales, hiperplasia del estroma velloso y vasos prominentes irregularmente engrosados, lumen estrecho y trombosis frecuente, no se observa hiperplasia del epitelio trofoblástico 2.
En 1991, Moscoso realizó la primera publicación y la llamó “anomalía vascular placentaria con hiperplasia difusa del mesénquima de las vellosidades primarias” 3, Lage JM le dio el término de “placentomegalia con hidrops masivo de las vellosidades coriales primarias, contenido de ADN diploide y onfalocele, posible asociación con el Sindrome de Beckwith-Wiederman (BWS)” 4.
La incidencia exacta de la DMP es desconocida. Arizawa y Nakayama revisaron 30 758 placentas y en 58 casos diagnosticados como mola completa (MHC), mola parcial (MHP) o relacionadas a BWS, encontraron 15 casos, 7 pertenecían a su hospital, con una incidencia de 0,02% 5; Zeng X en 95 595 placentas examinadas encontró 2 casos de DMP 6.
El feto generalmente es de sexo femenino 2 con una relación (F/M) que va de 3,6/1 7 a 8/1 8. En la mayoría de casos es diploide y normal genotípica 9 y fenotípicamente 2, pero se reporta algunos con anormalidades cromosomiales 7,10 como sindrome de Klinefelter 47,XXY, trisomía 13 o triploidía 69,XXX. Se asocia frecuentemente al Sindrome de Beckwith-Wiedemann 1,2,7, Nayeri 8 encontró 19% en 64 casos, Cohen 7 23% (15/66) y Pham 11 15 en 82 casos. También se han reportado otras malformaciones como cardiopatía congénita 7,12,13, arco aórtico interrumpido 7, mal rotación intestinal, hidronefrosis 14,15, onfalocele 4,14,15, arteria umbilical única 11,12, hiperplasia adrenal congénita 16 y tumores congénitos como el hamartoma mesenquimal hepático 17-20 que es el más frecuentemente reportado, hemangioma hepático y dérmico, hemangioendotelioma 13, hepatoblastoma 13, hamartoma pulmonar 18, cistoadenoma seroso de pancreas 21. Jimbo reportó un caso con sindrome de coagulación intravascular diseminada (CID) 22.
El cuadro clínico en la DMP es inespecífico, la mayoría se diagnostican por el hallazgo ecográfico o por el estudio de la placenta; los niveles de βHGC son normales 2, pero el alfa fetoproteína (AFP) puede estar elevada. Nayeri 8, en 61 casos revisados, solo el 9% de los embarazos afectados por PMD cursaron sin complicaciones maternas o perinatales. Pham 11 en un metanálisis, excluyendo fetos con BWS, encontró RCIU en el 50%, muerte perinatal 42%, o pretérmino en un 50%. Las complicaciones maternas fueron oligohidramnios 7, placenta previa, pre-eclampsia 23, hipertensión gestacional y síndrome HELLP.
REPORTE DE CASOS
Se presenta el reporte de cuatro casos de displasia mesenquimal placentaria, cuyos hallazgos clínico patológicos se muestran y resumen en la tabla 1.
Tabla 1. Hallazgos clínico patológicos de cuatro casos de displasia mesenquimal placentaria.
NE= No evaluable, MAC= Método anticonceptivo, NSE = no se encontraron datos.
Caso 1
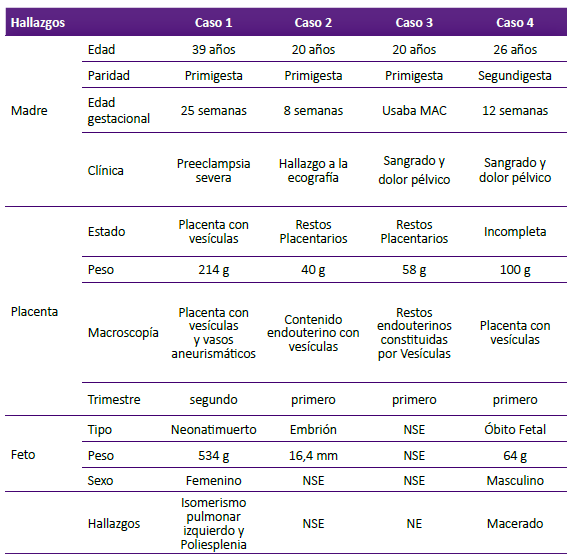
Primigesta de 39 años de edad, con 25 semanas de gestación, ingresó al hospital por un cuadro de preclampsia severa y evolucionó con compromiso renal, hipertensión 170/110 y cefalea. A la ecografía se diagnosticó MHP (Figura 1a). A la cesárea se obtuvo neonato prematuro, de sexo femenino que vivió 1 hora, pesó 534 g, con una talla de 28,3 cm. La placenta incompleta pesó 214 g; en la cara fetal (Figura 1b) presentó vasos tortuosos prominentes y a nivel de la cara materna (Figura 1c) se encontró numerosas vesículas distribuidas entre el parénquima normal, la mayor de las cuales midió 2,5 cm de diámetro. La microscopía reveló vellosidades coriales troncales hidrópicas con vasos arteriolares de pared irregularmente gruesa, algunos trombosados (Figura 1d), el estroma velloso hiperplásico (Figura 1e) y cisternas de diversos tamaños (Figura 1f). No se observó hiperplasia del epitelio trofoblástico. La autopsia neonatal (Figura 1g), reveló Isomerismo pulmonar izquierdo (Figura 1h) y bazo accesorio (Figura 1i).
Caso 2
Primigesta de 20 años, a las 8 semanas de gestación acude al centro de salud para control prenatal. En la ecografía se encontró saco gestacional con embrión de 16,4 mm sin latido cardiaco y sitio placentario compatible con MHP (Figura 2a); fue referida a un hospital de mayor complejidad. Exámenes auxiliares, βHCG = 9139 UI; otros exámenes de laboratorio dentro de límites normales. Se le realizó AMEU y legrado uterino. En patología se recibió contenido endouterino que medía 12x5x1,2 cm de diámetros mayores, donde se distinguía vesículas de diversos tamaños, el mayor midió 1,6 cm (Figura 2b y Figura 2c). A la microscopía, vellosidades troncales con degeneración hidrópica, grandes cisternas y vasos tortuosos, algunos con trombosis (Figura 2d).

Caso 3
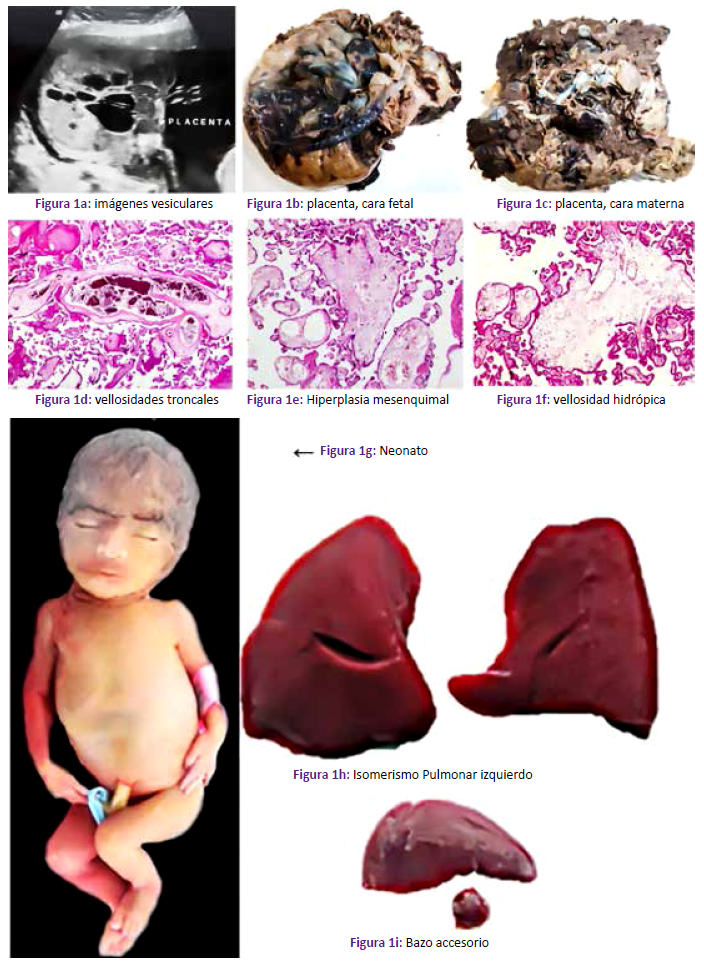
Primigesta de 20 años, acude por sangrado vaginal escaso de un día de evolución y dolor pélvico que se intensificó; la ecografía mostró lesiones diagnosticadas como “embarazo molar” (Figura 3a). Usaba métodos anticonceptivos y no menstruaba desde hace 5 meses. Se realizó legrado uterino. En patología se recibió contenido endouterino constituido en su mayor parte por vesículas a manera de “racimo de uvas” (Figura 3b y Figura 3c). A la microscopía se visualizó vellosidades troncales hidrópicas, con amplias cisternas, gran proliferación mesenquimal y vasos de paredes irregularmente engrosadas (Figuras 3d a 3g).
Caso 4
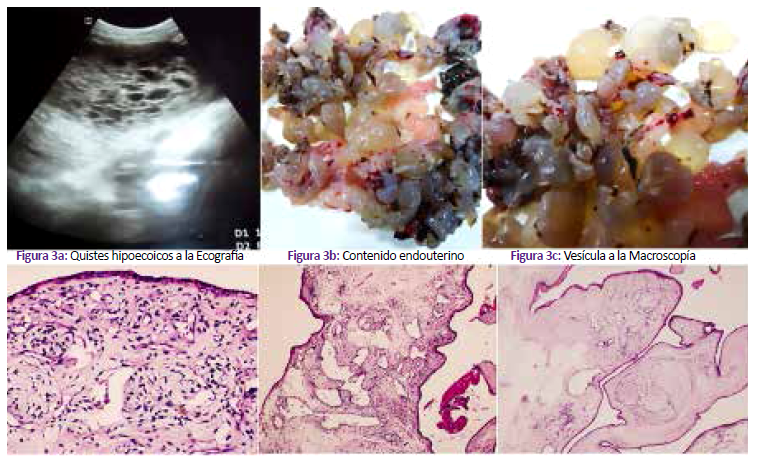
Segundigesta de 26 años, acude a emergencia por un cuadro de dolor pélvico y sangrado vaginal; a la ecografía se encuentró óbito fetal y placenta catalogada de MHP. Los exámenes de laboratorio estuvieron dentro de límites normales. En patología la placenta midió 10x10x2 cm, pesó 100 g; en la cara materna se halló múltiples vesículas de 0,5 a 2 cm de diámetro distribuidas dentro del parénquima normal (Figura 4a y Figura 4b). La histopatología mostró vellosidades troncales con degeneración hidrópica, hiperplasia del estroma y vasos con paredes engrosadas (Figuras 4c a 4e). El feto de 64 g estaba macerado y a la autopsia no se encontraron malformaciones congénitas (Figura 4f).
DISCUSIÓN
La DMP es una entidad de rara prevalencia, se menciona entre el 0,02% 5 en placentas examinadas en patología. Esta baja frecuencia puede deberse al subreporte o su falta de identificación porque la DMP aún no ha obtenido un reconocimiento amplio como entidad específica24,25; así, Paradinas 2 en el 2001 publicó una revisión, en un periodo de 25 años, de 7560 láminas histológicas reportadas como enfermedad trofoblástica y encontró 15 casos de DMP. Alvarez 25, de 1986 al 2017, reunió 104 casos de DMP, donde, 47(45,1%) fueron publicados en los últimos 5 años y según la especialidad del primer autor 54,8% eran ginecólogos, 19,2% anatomopatólogos, 9,6% pediatras, 5,7% radiólogos y más del 10% de otras especialidades.
La DMP se diferencia clínica, histológica y genéticamente de la MHP, del embarazo gemelar dicoriónico con feto normal y MHC (mola gemelar), del mosaicismo confinado a la placenta y de los abortos espontáneos con degeneración hidrópica 1,8, por ello el diagnóstico diferencial debe precisarse porque los pronósticos maternos y fetales difieren en cada caso. El feto generalmente presenta un cariotipo femenino normal 46XX 2,5,9, su asociación más frecuente es el sindrome de Beckwith-Wiedemann en un 25-30% 24, con una morbimortalidad fetal significativa. Pham 11, en un metanálisis, encontró en fetos sin BWS, una tasa de RCIU del 50%, muchos pesaron menos del percentil 5 y la tasa de muerte fetal y neonatal de 36% y 7% respectivamente.
La patología en la DMP muestra una placentomegalia, vasos sinuosos y muy engrosados, a menudo trombosados 26, presencia de vesículas de diferentes tamaños dispersas entre el parénquima normal; la microscopía revela vellosidades troncales hidrópicas con cisternas irregulares, estroma muy hiperplásico, vasos con paredes irregularmente gruesas, lumen estrecho y trombosis; no se observa hiperplasia del epitelio trofoblástico y las vellosidades terminales son normales. A la inmuhohistoquímica el estroma velloso de las vellosidades troncales anormales se tiñe para desmina y vimentina a igual que las vellosidades coriales normales pero son negativas para la actina del músculo liso 26.
La citogenética de la MHC presenta un complemento cromosómico diploide androgenético 26,27 y la MHP, triploide diándrico 26; Kaiser-Roger 27 en el 2006 publicó un estudio citogenético en dos casos de DMP y encontró un mosaicismo biparental androgénico, que comprometía el mesénquima y los vasos de estas vellosidades anormales, más no el epitelio trofoblástico. Este mosaicismo androgenético/biparental en la DMP, sería una explicación de su etiología.
El gen p57KIP2 ubicado en el cromosoma 11p15.5, tiene impronta genómica paterna y una expresión materna, la inmunotinción positiva de su proteína p57 indica la presencia de un alelo materno funcional, la MHC que carece de un genoma materno es negativa a la p57, en la MHP la p57 es positiva 28. En la DMP la tinción de nucleos del citotrofoblasto velloso de la p57 es positiva, pero negativa en los nucleos de células del estroma y vasos de las vellosidades anormales 28,29; la decidua es positiva 29 y se utiliza como control interno. La pérdida de tinción de p57 en la DMP podría tener valor diagnóstico para ayudar a distinguirla de las otras lesiones molares.
Los cuatro casos de DMP que reportamos cursaron con muerte perinatal, todas fueron diagnosticadas al ultrasonido como “mola”, tres mujeres fueron primigestas, la de mayor edad presentó pre eclampsia severa y en una fue un hallazgo en el control prenatal. La patología, en tres casos, las vesículas estaban dispersas entre el parénquima normal y en uno, toda la muestra estaba constituida por grandes vesículas, podría deberse a una edad gestacional temprana de la DMP cuando la mayor parte de las vellosidades son de tipo troncal. La microscopía en todos fue la clásica que se describe en la DMP, en el estudio fetal hallamos una malformación congénita no publicada a la fecha, el isomerismo pulmonar izquierdo con poliesplenia.
La DMP debe ser considerada como una probabilidad diagnóstica cuando se encuentre a la ecografía imágenes de aspecto molar con presencia fetal y HCG normal; el diagnóstico definitivo es microscópico. El hallazgo en la ecografía prenatal de quistes hipoecoicos en la placenta con feto vivo justifica un análisis citogenético para diferenciar entre PMD y un embarazo molar con un feto normal coexistente y evitar la culminación innecesaria. La inmunohistoquímica con p57, sería una valiosa y económica herramienta auxiliar para distinguir las diversas lesiones placentarias con degeneración molar y presencia de feto. Se debe publicar todo caso de DMP a fin de tener mayor conocimiento de esta entidad tan poco reconocida.

