











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCION
La encefalomiopatía mitocondrial, acidosis láctica y episodios stroke-like (episodios similares a enfermedad cerebrovascular), conocida como MELAS por su acrónimo en inglés (Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes), es un trastorno multisistémico progresivo causado por variantes genéticas en el ADN mitocondrial, considerada una enfermedad rara y huérfana 1-3. En 1982, se acuñó la denominación MELA para referirse a la Encefalopatía mitocondrial con acidosis láctica 4. En 1984, se presentaron las características clínicas distintivas de tres síndromes mitocondriales y se introduce por primera vez el acrónimo “MELAS” como un síndrome distintivo que incluye además a los episodios stroke-like 5.
Los pocos estudios de prevalencia región-específicos, sugieren que la variante m.3243A> G en el gen MT-TL1, sería la más frecuentemente asociada a MELAS. En la región europea se estimó una frecuencia de esta variante entre 16-236/ 100000 habitantes adultos y se ha descrito una prevalencia en población infantil de 18.4/ 100000 6,7,9. En Japón se ha estimado una prevalencia global, incluyendo diversas variantes, en 0.18/100000, siendo 0.50 (IC 95%, 0.41-0.59)/ 100,000 en menores de 18 años y 0.12 (IC 95%, 0.10- 0.14)/100,000 en población adulta (10). En otros países, como España, se han descrito casos sin estimaciones poblacionales 11.
MELAS en Latinoamérica
No hemos encontrado estudios de prevalencia de MELAS en Latinoamérica, es por ello que realizamos una estrategia de búsqueda sistemática de las bases de datos LILACS/SciELO, PubMed/Medline y Scopus desde 1990 hasta el 30 de junio de 2021. La estrategia de búsqueda consistió en palabras clave como "MELAS Syndrome" y "MELAS", y otros términos relacionados ("m.3243A>G", “stroke like episodes”), restringiendo la búsqueda a países de Latinoamérica y el Caribe (Latin America, Hispanic, Central America, South America).
Los criterios de inclusión fueron: 1) reportes de casos o series de casos de Síndrome de MELAS en Latinoamérica; 2) publicaciones que describieron datos sociodemográficos y clínicos de los pacientes (incluyendo datos individuales o agregados); 3) diagnóstico genético de la enfermedad y 4) disponibilidad del texto completo o resúmenes en inglés o español. Se utilizó el programa Rayyan para seleccionar los estudios y eliminar duplicados. Posteriormente, dos autores (MVT & WAQ) seleccionaron de forma independiente los informes y extrajeron los datos en Microsoft Excel.
Evaluación de la calidad
Utilizamos la herramienta propuesta por Murad et al.(12) Esta herramienta evalúa la calidad metodológica de los informes de casos/series evaluando 8 preguntas sobre cuatro dominios. Dado que nuestra revisión no se centra en los casos de eventos adversos, seleccionamos sólo 6 preguntas para nuestra evaluación: 1, 2, 3, 4, 7 y 8.
Extracción de datos y síntesis narrativa
Los datos fueron extraídos de forma independiente por dos autores (MVT y WAQ). Se extrajo la siguiente información: país, nombre del primer autor, año de publicación, sexo, edad de inicio, síntomas iniciales, cuadro clínico, resultados de ácido láctico, resultados de tomografía y resonancia magnética cerebral, biopsia muscular y estudio genético. Se describen las variables categóricas utilizando frecuencias y porcentajes, mientras que las numéricas como medianas y rangos intercuartílicos. Se utilizó el software Stata 17 (StataCorp. 2021. TX, USA). Se realizó un enfoque narrativo para sintetizar los estudios incluidos.
RESULTADOS
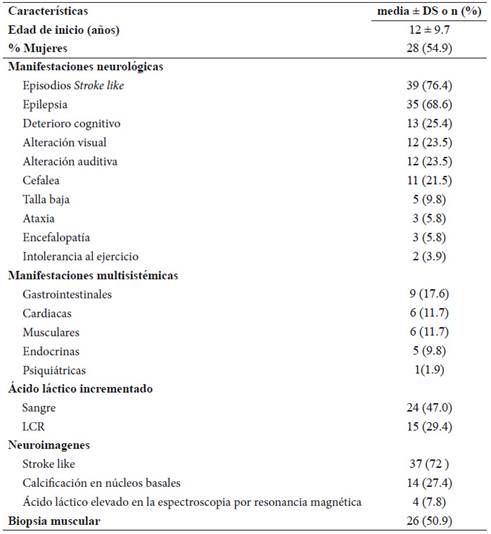
De los 966 estudios de la búsqueda global de las bases de datos utilizadas, únicamente 19 reportes de caso y series de caso fueron seleccionados. Un total de 51 casos con diagnóstico clínico de MELAS, 12 adultos y 39 niños, fueron descritos en las publicaciones incluidas; de estos, únicamente 42 (82%) describen el estudio genético: 40 corresponden a la variante m.3243A>G, 1 caso a la variante 13513 G>A y 1 caso a la variante 3271T>C. La edad media de inicio de la enfermedad es 12 ± 9,7años, con un predominio en el sexo femenino (Gráfico 1). Las manifestaciones neurológicas más frecuentes fueron los episodios de stroke like y epilepsia. Los episodios stroke like se presentaron en 66% (8/12) de adultos y 79 % (31/39) en niños. La epilepsia se presentó en 42 % de adultos (5/12) y 77% en niños (30/39) En neuroimágenes se evidenció principalmente lesiones isquémicas tipo stroke like y calcificación de núcleos basales. Lactacidemia y una biopsia positiva para fibras rojo-rasgadas fueron descritas en aproximadamente el 50 % de casos. Estas características se resumen en la Tabla 1.
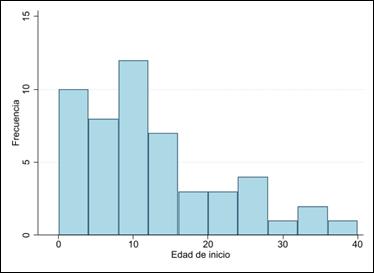
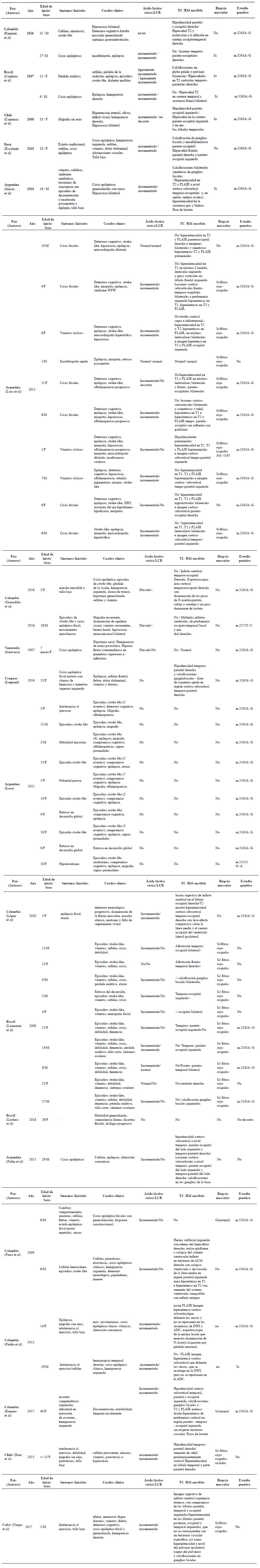
La revisión de la literatura en Latinoamérica mostró una cantidad reducida de casos de MELAS publicados. De estos no todos son casos confirmados con diagnóstico genético, siendo la mayoría de los reportes relacionados a la variante m.3243A>G, lo cual limita el conocimiento de la distribución de las variantes relacionadas a esta enfermedad en Latinoamérica; sin embargo, se encontraron similitudes en cuanto a las características clínicas y diagnósticas (Figura 1 y Anexo 1).
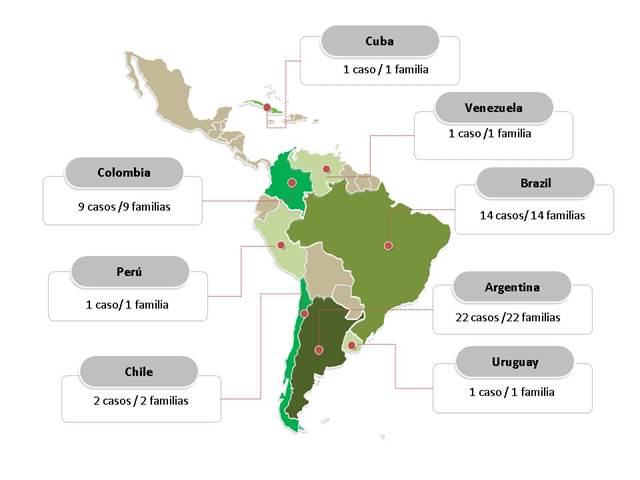
Figura 1 Distribución de casos de MELAS publicados en Latinoamérica. Los casos con diagnóstico genético descritos en Colombia fueron 26, sin embargo solo 9 de ellos (probandos de cada familia) tiene información clínica asociada.
ETIOPATOGENIA
La fisiopatología de la enfermedad no está completamente esclarecida; sin embargo, parte del fenotipo de la enfermedad puede ser explicada parcialmente por varios mecanismos que incluyen la producción de energía mitocondrial alterada, angiopatía de la microvasculatura y deficiencia de óxido nítrico 2.
Alteraciones genéticas
La enfermedad de MELAS es causada por variantes patogénicas en el ADN mitocondrial. En 1990 se describió por primera vez la variante m.3243A> G en el gen MT-TL1, que representa la causa genética de MELAS hasta en el 80% de los casos. Actualmente, se han descrito hasta 34 variantes patogénicas asociadas a MELAS en el gen MT-TL1 y otros genes mitocondriales implicados 1.
Deficiencia energética y angiopatía
Se ha visto que la variante m.3243A> G produce una traducción mitocondrial alterada con una disminución de la síntesis de proteínas mitocondriales afectando a las subunidades del complejo de la cadena trasportadora de electrones (CTE), y esto da como resultado una producción de energía mitocondrial deficiente 13,14. La insuficiente generación de ATP para satisfacer las necesidades energéticas da como resultado la disfunción multiorgánica y una estimulación en la proliferación mitocondrial. La angiopatía debido a la proliferación mitocondrial en el músculo liso y las células endoteliales de los vasos sanguíneos pequeños conduce a una perfusión sanguínea alterada en la microvasculatura que contribuye significativamente a las complicaciones, específicamente a los episodios stroke like 2.
Deficiencia de óxido nítrico (ON)
Se ha planteado que la deficiencia de ON ocurre en el síndrome MELAS y puede contribuir significativamente a sus complicaciones 15,16. El ON se forma a partir de la arginina a través de la enzima óxido nítrico sintasa, que cataliza la conversión de la arginina en citrulina. Tanto la citrulina como la arginina se consideran donantes de ON. El ON producido por el endotelio vascular desempeña un papel importante en la relajación del músculo liso vascular necesaria para mantener la permeabilidad de los vasos sanguíneos pequeños. Por tanto, la deficiencia de ON resulta en una alteración de la perfusión sanguínea en la microvasculatura de diferentes órganos que puede contribuir a la patogénesis de varias complicaciones 17. Se cree que la deficiencia de ON en el síndrome de MELAS es de origen multifactorial. La proliferación mitocondrial en las células endoteliales vasculares puede resultar en una función endotelial alterada y una síntesis de ON alterada. La sobreproducción de especies reactivas de oxígeno (estrés oxidativo) resultante de la alteración del CTE también puede disminuir la producción de ON18. La lactacidemia es el resultado de la incapacidad de las mitocondrias disfuncionales para oxidar adecuadamente la glucosa, lo que lleva a la acumulación de piruvato y derivación de piruvato a lactato 19. Además, la hipoperfusión puede provocar acidosis láctica debido a la disminución del suministro de oxígeno a los tejidos periféricos y un cambio a la glucólisis anaeróbica. La deficiencia de ON en el síndrome de MELAS puede resultar en una disminución de la perfusión sanguínea y, por lo tanto, puede agravar la acidosis láctica 17.
CUADRO CLÍNICO
La enfermedad de MELAS es un trastorno multisistémico que se presenta en todos los grupos etarios. La mayoría de los casos de MELAS se encuentran entre 2-40 años, con más de la mitad de los casos con inicio de síntomas antes de los 20 años 2 . Sin embargo, existen casos de presentación tardía después de los 40 años (20). La variabilidad fenotípica en MELAS se explica por: 1) la heteroplasmia, coexistencia de ADN mitocondrial normal y mutado. 2) La distribución variable del ADN mitondrial mutado en los tejidos y 3) la respuesta variable de cada tejido corporal al estrés oxidativo.
Las manifestaciones clínicas de MELAS se describen en la Tabla 2. Las formas juveniles y las del adulto cursan con episodios similares a enfermedad cerebrovascular o stroke-like, cefaleas recurrentes y epilepsia 2,10. Hirano observó 6 manifestaciones cardinales: stroke-like, crisis epilépticas, acidosis láctica, fibras rojo rasgadas, intolerancia al ejercicio y comienzo de los síntomas antes de los 40 años en más del 90% de los 110 casos reportados 21. En las formas juveniles de MELAS predominan la baja estatura y el retraso en el crecimiento, mientras que en la forma adulta diabetes mellitus y pérdida auditiva 10.
Tabla 2 Manifestaciones iniciales y cuadro clínico en MELAS
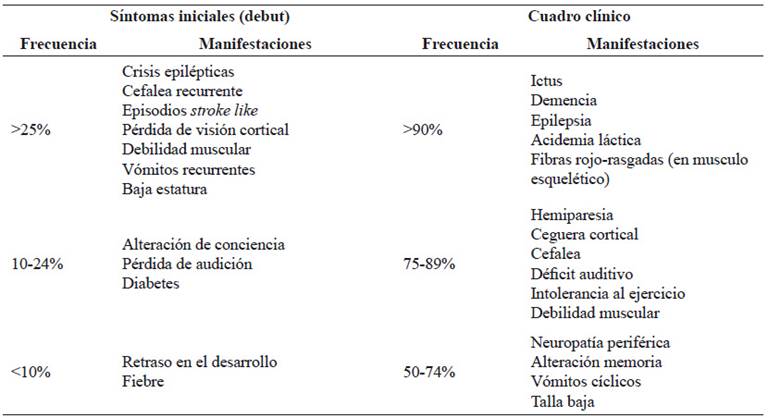
Tomado y adaptado de El-Hattab et al, 2015 (2)
Los episodios stroke-like son uno de los síntomas más frecuentes de la enfermedad. Son episodios habitualmente reversibles de afasia, pérdida de visión cortical, debilidad motora, cefalea, crisis o alteración del estado mental; sin embargo, se han descrito formas progresivas de déficit neurológico19).
La miopatía es frecuente en pacientes enfermedad mitocondrial incluyendo MELAS 22. Esta se presenta como intolerancia al ejercicio en un 73-100% y la debilidad muscular proximal en 42-89% 10,19,21.
Las crisis epilépticas se presentan hasta en un 71-96%, pueden ser focales o generalizadas, episodios aislados o en el contexto de un episodio stroke-like 23. La cefalea recurrente se presenta entre 54-91% 10,19,21. La cefalea suele ser más intensa al asociarse a episodios de stroke-like 24.La neuropatía periférica es otra manifestación común que ocurre en 22-77%. La neuropatía axonal o axonal mixta y desmielinizante es el tipo más común 25,26. La demencia suele presentarse entre 40-90% 10,21. Esta afecta principalmente el lenguaje, la memoria y percepción visuoespacial19.
La enfermedad de MELAS habitualmente se asocia a varios síntomas extraneurológicos. Se ha observado compromiso cardíaco tanto miocardiopatía dilatada como hipertrófica; sin embargo, la más típica es una miocardiopatía hipertrofia concéntrica no obstructiva 19. Se han descrito también anomalías en la conducción cardiaca como el síndrome de Wolff- Parkinson-White que se presenta en 13-27%. 27. A nivel gastrointestinal, se ha descrito vómitos recurrentes hasta en un 77% de casos; así como diarrea, estreñimiento, dismotilidad gástrica 19,21,28. La presencia de diabetes en MELAS se ha descrito hasta en 33% de los casos 10,19. Algunos estudios sugieren una asociación entre la variante m.3243A> G y una mayor ocurrencia de diabetes mellitus 29). Se postula que el fracaso en la secreción de insulina por las células β pancreáticas sería el mecanismo desencadenante de diabetes, posiblemente debido a un fallo de energía y la dependencia de estas células de bombas de sodio-potasio impulsadas por ATP 19,29. En la diabetes mellitus no insulino dependiente se debe evitar el uso de metformina debido a que éste fármaco causa acidosis láctica 19. Se ha descrito síntomas psiquiátricos asociados a MELAS incluyendo trastornos del ánimo, psicosis y ansiedad 30.
DIAGNÓSTICO
Diagnóstico clínico de MELAS
El diagnóstico clínico de MELAS ha variado en el tiempo, desde que se describió por primera vez. Se han publicado dos conjuntos de criterios, los primeros fueron dados por Hirano en 1992 al analizar 69 reportes de caso 4. En 2012 se plantearon nuevos criterios diagnósticos dados por el comité de estudio de MELAS en Japón 10), que establece el diagnóstico clínico definitivo de MELAS con dos criterios pertenecientes a la categoría A y dos criterios de la categoría B; y sospecha de MELAS con un criterio de la categoría A y dos criterios de la categoría B (Tabla 3).

Estudios paraclínicos de apoyo diagnóstico
Neuroimágenes
El uso de imágenes en MELAS tiene mayor utilidad durante los episodios agudos de la enfermedad. La tomografía cerebral ha sido muy utilizada inicialmente en los cuadros agudos de stroke-like. Los hallazgos más frecuentes en neuroimagen en MELAS incluyen la calcificación de núcleos basales bilaterales y tálamos. Se han descrito también, áreas corticales de hipodensidad localizadas en uno o ambos polos occipitales no confinadas a un territorio vascular; la afectación es habitualmente cortical 31).
Las imágenes por resonancia magnética ponderadas por difusión (DWI) son útiles en MELAS. A pesar de que habitualmente son utilizadas para evidenciar lesiones isquémicas, también resultan útiles en otras lesiones no isquémicas que pueden mimetizar un infarto como las patologías por enfermedad mitocondrial. A diferencia de un infarto habitual, en los episodios de stroke-like se plantea una superposición temporal del edema citotóxico inicial en la fase aguda y el edema vasogénico en la fase subaguda a crónica de las lesiones, lo cual estaría representado por una disminución del coeficiente de difusión aparente (ADC) inicial y posteriormente un aumento de señal en ADC 32. Las lesiones observadas en resonancia magnética no se corresponden con un territorio vascular, estas tienen una localización cortical. A pesar que las lesiones stroke like pueden presentarse en cualquier localización, suelen ser más frecuentes en lóbulos parietales, temporales y occipitales. Pueden existir cambios en sustancia gris profunda (núcleo caudado, globo pálido, putamen, núcleo pulvinar del tálamo y núcleo dentado del cerebelo). Se han descrito también microcalcificaciones y depósitos de calcio y hierro a nivel de núcleos basales. La espectroscopia por resonancia magnética detecta una amplia gama de metabolitos que podrían modificarse en enfermedades metabólicas, describiéndose que la disminución de N acetil aspartato (NAA) asociado a un incremento en el pico de lactato sería útil en el diagnóstico de pacientes con MELAS 32.
Electromiografía y velocidad de conducción: En pacientes con enfermedades mitocondriales se ha descrito neuropatía axonal, desmielinizante o mixta. No se ha determinado si la neuropatía es causada directamente por disfunción mitocondrial o por mecanismos patogénicos secundarios a la deficiencia de la cadena respiratoria. En la mayoría de los estudios en pacientes con síndrome de MELAS se ha encontrado que la polineuropatía es de tipo axonal, siendo los hombres los más susceptibles a presentarla. 18,19.
Ácido láctico: La presencia de hiperlactacidemia se considera un síntoma cardinal, aunque inespecífico de MELAS. Los valores de lactato se pueden encontrar elevados en sangre y en LCR en la mayoría de los casos 1).
Biopsia muscular: El examen histológico muestra fibras musculares vacuoladas dispersas con un borde transparente con la tinción hematoxilina-eosina. Con la tinción tricrómica de Gomori, las fibras musculares afectadas se ven como fibras rojo-rasgadas (FRR) debido a la proliferación mitocondrial por debajo de la membrana plasmática. Las FRR están presentes en muchas otras enfermedades mitocondriales; sin embargo, en MELAS estas se tiñen positivamente con tinción citocromo oxidasa (COX) a diferencia de otras enfermedades mitocondriales que no reaccionan con COX 1,19.
Estudio genético molecular
Las variantes patogénicas asociadas a MELAS (Tabla 4), se detectan en ADN mitocondrial (MtDNA) extraído de leucocitos de individuos afectados. La heteroplasmia, característica de las enfermedades mitocondriales, puede generar una distribución variable del mtDNA, por lo que las variantes podrían solo detectarse en otros tejidos como mucosa bucal, sedimento urinario o mucho más consistentemente en musculo esquelético 22,33. La recomendación es realizar estudio genético en ADN mitocondrial extraido de al menos dos diferentes tejidos.
Tabla 4 Genes y variantes genotípicas asociadas a MELAS

Tomado y adaptado de El-Hattab et al, 2018 (1)
La aproximación diagnóstica molecular más adecuada dependerá de lo característico del fenotipo de MELAS. Si el caso tiene signos clínicos típicos de MELAS, se recomienda estudio molecular de un gen-especifico, para las variantes m.3243A> G, y en caso negativo para las variantes m.3271T> C, 3252A> G (gen MT-TL1) y m.13513G> A (gen MT-ND5); también se puede realizar panel de genes mitocondriales o secuenciación completa de genoma mitocondrial. En los casos de fenotipos no característicos o complejos se recomienda estudios basados en panel de genes, exoma o genoma completo 1.
Diagnóstico diferencial
El cuadro clínico de MELAS se superpone con diversas enfermedades mitocondriales como síndrome de Kearns Sayre, MERRF y síndrome de Leigh.
El diagnóstico diferencial de los episodios de stroke like en personas jóvenes incluye: enfermedad cardíaca, enfermedades carotídeas o vertebrales, enfermedad de células falciformes, vasculopatías, discrasias de lipoproteínas, trombosis venosa, enfermedad de moyamoya, migraña complicada, enfermedad de Fabry y homocistinuria.
También se ha reportado fenotipos similares a MELAS asociados a variantes en genes nucleares como MRM234 , FASTKD235 y POLG36.
MANEJO INTEGRAL Y MULTIDISCIPLINARIO
Todo paciente con enfermedad de MELAS debe tener un abordaje integral y multidisciplinario. A la fecha no hay tratamiento farmacológico específico para MELAS. Sin embargo, se utilizan algunos fármacos antioxidantes como Coenzima Q10, L-Carnitina y creatina. Actualmente hay 3 ensayos clínicos en fase I y II que se encuentran evaluando intervenciones como L-Citrulina y Glutamina oral para pacientes con MELAS (www.clinicaltrials.gov).
La coenzima Q es utilizada en el síndrome de MELAS ya que facilita la transferencia de electrones del complejo I y II al complejo III de la CTE y la estabiliza al proveer protección antioxidante. Algunos estudios han demostrado un efecto beneficioso en la debilidad muscular, fatigabilidad y niveles de lactato 37. La dosis recomendada en adultos es de 200 - 400 mg/día, con dosis máxima de 600 mg/día. La dosis pediátrica recomendada es 3.4-10 mg/día (nivel de evidencia bajo) 38.
La L-carnitina ha sido utilizada debido a que se ha descrito una deficiencia secundaria en algunos trastornos mitocondriales como MELAS, siendo su función el transporte de ácidos grasos de cadena larga a la matriz mitocondrial para la β-oxidación; sin embargo, actualmente no se recomienda su uso prolongado debido al riesgo de ateroesclerosis. La L-carnitina se puede administrar en dosis de 3 g diarios en tres dosis divididas para adultos y 100 mg / kg / día para niños en tres dosis divididas ( nivel de evidencia bajo) 38.
La creatina monohidrato también se ha utilizado en el tratamiento de MELAS aunque con poca evidencia. Se ha documentado cierta mejoría en la fuerza muscular luego de actividad aeróbica y anaeróbica en pacientes con trastornos mitocondriales, así como también una leve mejoría en las crisis epilépticas. Aunque se debe tener en cuenta sus efectos adversos en la función renal, sobretodo en pacientes con la variante m.3243A> G. En adultos se recoemienda una dosis de carga de 20 g/día por 2 semanas y una dosis de mantenimiento de 2-5 g/día. En niños se utiliza a una dosis de 80-350 mg/kg/día 38 (nivel de evidencia bajo)
Para el tratamiento de los episodios stroke like se recomienda L-arginina. Durante el episodio agudo se recomienda administrar un bolo de arginina intravenosa (500 mg / kg para niños o 10 g/m2 de superficie corporal para adultos). L-arginina se evaluó en un ensayo clínico abierto de fase 3, en el cual la presentación oral demostró eficacia para extender la fase interictal y disminuir la incidencia y severidad del episodio stroke like. L-arginina intravenosa también mejoró las tasas de cefalea, náuseas y vómitos, alteración de la conciencia y alteración visual (nivel de evidencia moderado) 39. La taurina, un β-aminoácido que está ausente en células derivadas de pacientes con MELAS asociadas a la variante m.3243A>G o m.3271T>C, es otro tratamiento que ha sido evaluado en un pequeño ensayo clínico de fase 3, a una dosis de 9 y 12 g día, que demostró eficacia en la reducción de la recurrencia de episodios stroke like (nivel de evidencia moderado) 40.
Para los episodios de crisis epiléptica se recomienda el manejo habitual de anticonvulsivantes, evitando el uso de ácido valproico, debido a que puede alterar la membrana mitocondrial induciendo cambios de despolarización paroxística, así como la alteración en la actividad de bombeo de protones del complejo IV (citocromo c oxidasa) en la cadena respiratoria, estos cambios pueden precipitar aún más la sintomatología de los trastornos mitocondriales que de por si presentan una deficiencia de citocromo c oxidasa 41. Otros anticonvulsivantes no recomendados son: carbamazepina, fenitoína, fenobarbital y topiramato. Los anticonvulsivantes que han mostrado menor riesgo de toxicidad mitocondrial y adecuado balance riesgo beneficio son levetiracetam, zonisamide , lamotrigina y gabapentina. Asimismo, se recomienda evitar el uso o consumo de: metformina debido al mayor riesgo de desarrollar acidosis láctica, antibióticos aminoglucósidos y linezolid por el riesgo de precipitar la insuficiencia mitocondrial y una acidosis láctica42, cigarrillos y alcohol por el incremento del estrés oxidativo41.
Los pacientes MELAS deben tener evaluaciones por algunas especialidades médicas por riesgo de complicaciones sistémicas. Se recomienda manejo por cardiología para las complicaciones cardiacas como la miocardiopatía y el síndrome de Wolff-Parkinson-White; manejo endocrinológico para el control de la diabetes; evaluaciones por otorrinolaringología en los casos de hipoacusia, manejo nutricional para establecer una dieta adecuada en estos pacientes; evaluaciones psiquiátricas periódicas en pacientes que cursan con cuadros depresivos, así como asesoramiento genético individual y familiar adecuado 1.
El asesoramiento genético debe proporcionar información precisa sobre el pronóstico de las personas afectadas y el riesgo de recurrencia para otros miembros de la familia. La sesión tambien debe informar sobre consecuencias de la enfermedad en el paciente y la familia sobre opciones reproductivas y otras decisiones médicas y personales de vida. Se recomienda la evaluación y asesoramiento para todos los miembros de la familia, afectados y no afectados. En las enfermedad mitocondriales como MELAS, es importante comunicar a la familia que esta enfermedad se transmite por linea matrilineal; es por ello que la madre del paciente es usulmente portadora de la variante patogénica y puede tener o no tener síntomas, aunque hay casos de variantes genéticas mitocondriales de novo. Todos los hijos de una madre portadora de variantes patogénicas de MELAS heredan esta variante; sin embargo pueden o no desarrollar síntomas. Es por ello que la predicción de aparición de síntomas basado en pruebas genéticas no es posible 43.

