











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Las tubulopatías representan un grupo muy diverso de entidades con alteraciones en la función del túbulo renal, se diferencian las de causa primaria o de hereditarias de las causas secundarias, debido a que entre ellas el cuadro clínico, gravedad y pronostico son muy determinantes. 1
Las diferentes manifestaciones clínicas, su impacto en el crecimiento de los pacientes afectados y el riesgo de daño renal crónico, convierten a las tubulopatías en una patología importante en la edad pediátrica, que requiere conocimiento de fisiología de la función tubular en el personal médico que atiende a estos pacientes para su diagnóstico precoz y tratamiento oportuno. Actualmente debido al componente hereditario de algunas de las formas de tubulopatía, el estudio genético se ha convertido en una herramienta importante que nos permite obtener el diagnostico etiológico, definir el pronóstico a largo plazo y adoptar medidas preventivas para el curso de estas enfermedades.
Por este motivo se presenta el siguiente caso, considerado el primero en reportarse en nuestro país con el diagnostico de Hipomagnesemia Familiar con hipercalciuria y nefrocalcinosis. (HFHNC) 2, con estudio genético confirmado.
PRESENTACIÓN DEL CASO
Niña de 9 años, procedente de la provincia de Tocache, departamento de San Martin, Perú. Desde los 6 años presenta un cuadro caracterizado por dolor óseo, debilidad muscular, disuria y poliuria e historia de infección del tracto urinario (ITU) recurrente (3 episodios al año). Dos años antes de su ingreso presentó dificultad para la marcha acompañado de dolor óseo que progresó hasta la incapacidad total para la deambulación por lo que fue evaluada en el servicio de Neurología Pediátrica descartándose causa neurológica con el estudio de electromiografía. Veinte días antes del ingreso presentó cuadro clínico de infección urinaria fue evaluada en un hospital de su localidad donde evidenciaron exámenes de función renal alterados por lo cual fue referida a nuestro hospital.
Al ingreso la niña se encontraba en mal estado nutricional, con palidez marcada y debilidad muscular marcada a predominio de extremidades inferiores; los exámenes auxiliares mostraron enfermedad renal crónica estadio IV, anemia moderada, acidosis metabólica con anión gap normal, hipomagnesemia, hipocalcemia, hiperparatiroidismo, hipercalciuria y el examen de orina, leucocituria (tabla 1).
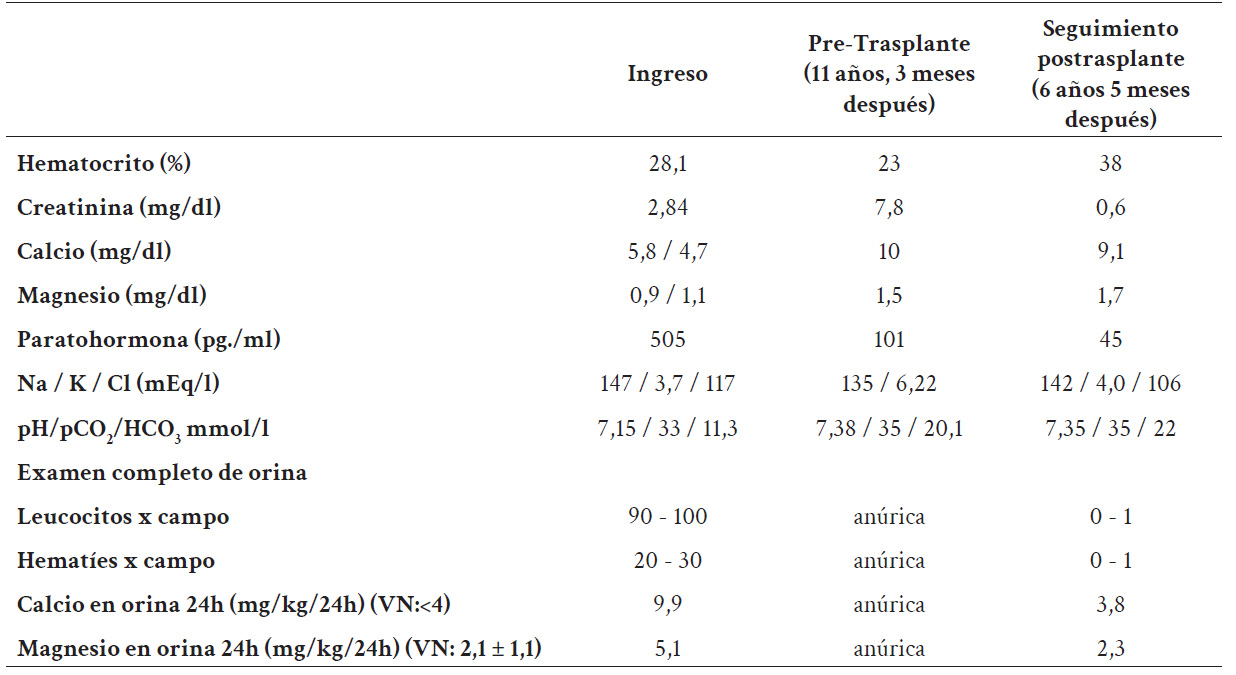
En la radiografía de abdomen simple se observó múltiples imágenes litiásicas compatible con nefrocalcinosis (figura 1). Durante la hospitalización presentó dos episodios de convulsiones y tetania relacionados a la hipocalcemia e hipomagnesemia severa. Por esta razón, se planteó el diagnóstico de hipocalcemia e hipomagnesemia sintomáticas, enfermedad renal crónica secundaria a tubulopatía primaria perdedora de calcio y magnesio, nefrocalcinosis y acidosis tubular renal, ITU recurrente y uropatía obstructiva por la presencia de cálculos en los uréteres. Como parte del manejo inicial se le realizó ureterolitotomía bilateral, y se inició terapia con citrato de potasio e hidroclorotiazida, magnesio vía oral y uso de profilaxis antimicrobiana para la ITU. Así mismo se encontró niveles de parathormona en 505 pg./ml motivo por el cual se inició calcitriol 0,25 µg cada 24 h vía oral.
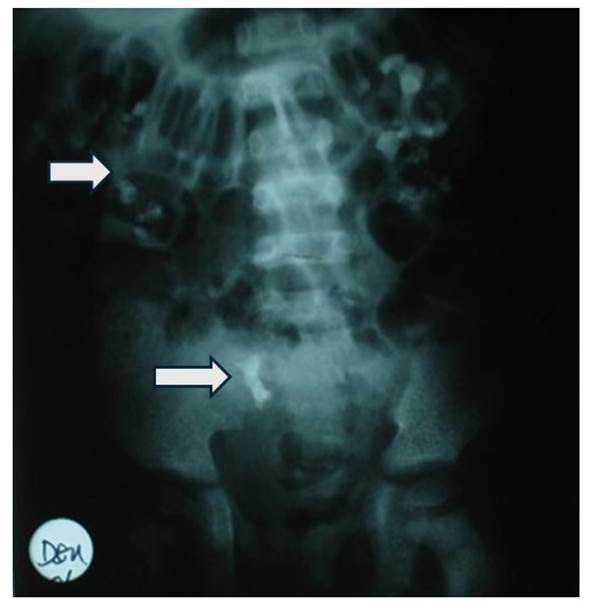
Figura 1 Radiografía de abdomen simple muestra múltiples imágenes radiopacas a nivel renal y vías urinarias (flechas), de tamaño variable.
La enfermedad renal (ERC) progreso a estadio V y a los 10 años progresó a ERC secundaria a uropatía obstructiva por litiasis bilateral y nefrocalcinosis ingresando al programa de diálisis peritoneal continua ambulatoria donde se mantuvo por 3 años y 6 meses, cursando con un episodio de peritonitis fúngica motivo por el cual ingresó a hemodiálisis crónica intermitente a los 13 años. Permaneció por 7 años con problemas de acceso vascular.
La paciente fue sometida a trasplante renal recibiendo un injerto renal de un donante cadavérico. La niña fue sometida a nefrectomía bilateral previo al trasplante renal. En el estudio de histocompatibilidad HLA presentó 2 alelos HLA-A*02 compatibles, recibió timoglobulina como terapia de inducción, tacrolimus, micofenolato de mofetilo y esteroides como terapia de mantenimiento con evolución favorable. Actualmente, la paciente mantiene inmunosupresión triple sin complicaciones. Su evolución ha sido favorable luego de 7 años de trasplante renal.
Debido al antecedente familiar de consanguinidad, antecedente de enfermedad renal en hermanos y cuadro compatible de tubulopatía como causa de ERC, se obtuvo una muestra de ADN genómico deshidratado, utilizando el método de precipitación en salado “Salting out”, en el laboratorio de Histocompatibilidad y Biología Molecular de la Unidad de Trasplante Renal del Hospital Cayetano Heredia y enviado al laboratorio de Investigación de Félix Claverie-Martin, PhD; Coordinador de RenalTube en Tenerife, España, para el estudio genético del trastorno tubular primario. El resultado fue una mutación patogénica en homocigosis c.446 G>A (p.R149Q) ubicado en el exón 3 del gen CLDN16, que permitió la confirmación diagnóstica etiológica de un caso de hipomagnesemia familiar con hipercalciuria y nefrocalcinosis (HFHNC), sin defectos oculares severos.
DISCUSIÓN
La hipomagnesemia familiar con hipercalciuria y nefrocalcinosis (HFHNC) es un trastorno tubular renal con un patrón autosómico recesivo caracterizado por pérdidas urinarias excesivas de magnesio y calcio, nefrocalcinosis bilateral e insuficiencia renal crónica progresiva en la infancia o la adolescencia.
HFHNC es una rara enfermedad de herencia autosómica recesiva, con aproximadamente 120 casos notificados en todo el mundo Es producida por mutaciones en las proteínas ubicadas en las zonas de uniones estrechas “tight junction” del espacio intercelular denominadas claudina-16 y claudina-19 que están codificadas por los genes CLDN16 y CLDN19, respectivamente. 3,7
Las claudinas son componentes clave del transporte paracelular, ubicadas en el segmento grueso ascendente del asa de Henle y desempeñan un papel importante en la regulación de la reabsorción de magnesio. Los canales para-celulares en la unión estrecha tienen propiedades de selectividad iónica similares a los canales transmembrana convencionales y debido a un gradiente de voltaje impulsa la reabsorción pasiva para celular de cationes divalentes como Mg2+ y Ca2+. 4
Las manifestaciones clínicas y alteraciones bioquímicas en los pacientes con HFHNC incluyen en el 100% de los casos la hipomagnesemia, hipercalciuria y nefrocalcinosis, pero además se ve que más del 80% puede presentar poliuria/polidipsia, incremento de la parathormona sérica, acidosis tubular renal distal incompleta e hipocitraturia. En menor proporción se han asociado manifestaciones como infecciones del tracto urinario, hiperuricemia, tetania y nefrolitiasis que acompañarían a la triada clásica según la evolución de la enfermedad (5.6). En el caso presentado la paciente tuvo síntomas inespecíficos durante casi tres años lo que no permitió una sospecha diagnóstica precoz y favoreció la progresión a un estadio avanzado de enfermedad renal crónica que se encontró al momento de su diagnóstico.
Debido al modo de herencia, una gran proporción de los pacientes afectados se origina en poblaciones con alta frecuencia de padres consanguíneos. Actualmente se han identificado un total de 68 mutaciones de CLDN16 diferentes y 19 mutaciones de CLDN19 3. Siendo en nuestro caso confirmado la consanguinidad de los padres, además de la afectación de una hermana con un cuadro similar pero aun sin la confirmación del estudio genético; debemos enfatizar que la mutación patogénica en homocigosis c.446 G>A (p.R149Q), encontrada en la paciente ha sido previamente descrita por Tajima et al. 8, en un niño japonés.
El único tratamiento actual para la enfermedad renal secundaria a HFHNC es el trasplante renal en la etapa terminal, ya que la terapia de apoyo con suplementos de magnesio y el uso de diuréticos tiazídicos solamente retrasan la aparición de ERCD 3. En la paciente se pudo llevar a cabo el trasplante renal en un momento de máxima gravedad sin tener aún la confirmación genética de su enfermedad de fondo la misma que recién se realizó dos años después durante su seguimiento.
La frecuencia de tubulopatías complejas en edad pediátrica está en incremento, son de presentación muy diversa y requieren un diagnóstico rápido debido a la gran importancia de su diagnóstico temprano y manejo oportuno para prevenir en primer lugar la afectación del crecimiento en el niño, en segundo lugar, la formación de cálculos y Nefrocalcinosis en edad temprana y así evitar el daño definitivo que requiera de terapias de reemplazo renal crónica.
En conclusión, el diagnostico etiológico obtenido en nuestra paciente nos evidencia la ocurrencia de tubulopatías hereditarias en nuestra población que requieren de estudios genéticos adicionales a nivel familiar para determinar la distribución de las mutaciones del gen CLDN16, de importancia en el tratamiento, pronóstico y prevención de nuevos casos mediante la consejería genética en las familias afectadas.

