










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Gastroenterología del Perú
versión impresa ISSN 1022-5129
Rev. gastroenterol. Perú v.32 n.1 Lima ene./mar. 2012
CONTRIBUCIÓN ESPECIAL
Infarto de Bazo y Hemoglobinopatía S en la Altura
Spleen infarction and S hemoglobinopathies S in the high altitude lands
Oscar E. Frisancho* y Carlos Ichiyanagui Rodríguez*
* Médico gastroenterólogo. Hospital Nacional Edgardo Rebagliati Martins, EsSALUD, Lima, Perú.
RESUMEN
La hemoglobinopatía S es un desorden hereditario resultado de una mutación en el gen beta-S que se expresa con la sustitución de un aminoácido en la cadena beta de globina. El problema se presenta cuando algún sujeto con hemoglobinopatía S se expone a la hipoxia de altura. La disminución de la saturación de oxígeno forma polímeros de Hb S que deforman al glóbulo rojo en forma de "media luna" (célula falciforme o drepanocito). Las células falciformes (rigidas y frágiles) tienden a adherirse a otros glóbulos rojos incrementando la viscosidad y estásis sanguínea, generando oclusión vascular e infarto en los tejidos. El bazo por su tipo de circulación es un órgano susceptible de la crisis falciforme. El infarto esplénico en la altura -en correspondencia a diferentes circunstancias- puede evolucionar en tres etapas: a) Infarto agudo (focal, no complicado), b) infarto masivo (compromiso de mas del 50% del parénquima) y c) infarto con disrupción capsular. El diagnóstico precoz es fundamental, permite la instauración oportuna de diversas medidas, especialmente una adecuada hidratación, oxigenación y rápida evacuación a localidades de menor altitud. Con estas medidas se atenua el fenómeno falciforme y algunos pacientes pueden superar este trance sin mayores complicaciones. El retardo diagnostico conlleva a tomar medidas que incluso pueden exacerbar la hipoxia tisular. Importantes poblaciones de raza negra y mestiza con ancestro africano viven en la costa peruana, 10% y 2% respectivamente tienen hemoglobinopatía S; sujetos de raza blanca con ancestro mediterráneo también pueden portar esta hemoglobina. Es indispensable difundir el conocimiento de esta entidad para que la tengan presente los médicos que laboran en regiones de altura; asimismo es primordial impulsar medidas preventivas para que los individuos con ancestro africano o mediterráneo conozcan su estatus sickle cell antes de viajar a lugares por encima de 2.500 m.
PALABRAS CLAVE: hemoglobina S, infarto de bazo, sickle cell portador.
ABSTRACT
The hemoglobin S is a consequence of the substitution of valine for glutamic acid at position 6 of beta globin chain. The problem arises when some individuals with Hb S is moved to the mountains and exposed to hypoxia. The decrease in oxygen saturation distorts the red blood cell with HbS-shaped crescent (sickle cell). Sickle cell (rigid and fragile) tends to adhere to the other red blood cells, generating a series of intravascular alterations that can lead to tissue ischemia or infarction. The spleen by type of movement and lack of lateral communications between the branches of the splenic artery was the most susceptible to sickle cell crisis. Splenic infarction at altitude corresponding to different circumstances can evolve in three stages: a) Acute (focal, uncomplicated), b) massive attack (more than 50% of parenchyma) and c) spontaneous rupture. Early diagnosis is crucial, allowing the quick and timely introduction of various measures, including adequate hydration and oxygenation continues until its evacuation to lower altitude locations. These measures would reduce the phenomenon of sickle and some patients may overcome this acute trance without major complications. The delay in diagnosis leads to action that can exacerbate tissue hypoxia and cause ischemia or infarction of various organs. A large population of black and mixed race of African descent living in the Peruvian coast, 10% and 2% respectively have hemoglobin S; Caucasian subjects with Mediterranean ancestry this hemoglobin also can carry. It is therefore essential to disseminate within the clinicians working in regions of high status and to thus prevent potentially fatal complications in patients with Hb S; is also essential to promote preventive measures for individuals with African or Mediterranean ancestry know their sickle cell status before traveling to places above 2,500 m.
KEY WORDS: hemoglobin S, spleen infarction, sickle cell trait.
INTRODUCCIÓN
La cordillera de los Andes determina una especial configuración geográfica de muchos países sudamericanos, en especial del Perú. En nuestro país existen muchas poblaciones encima de los tres mil metros de altura (sobre el nivel del mar); para llegar a ellos se impone la necesidad de recorrer caminos que en su mayoría atraviesan grandes alturas, a veces de más de 4,800 m. como Ticlio.
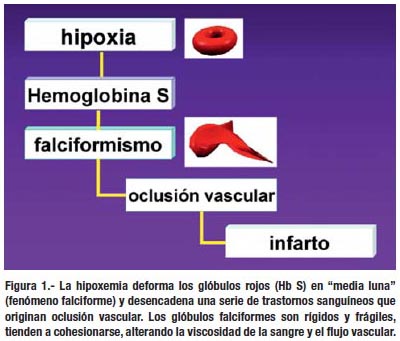
El problema se presenta cuando algún sujeto con hemoglobinopatía S se traslada a la sierra y se expone a la hipoxia ambiental, La hipoxemia provoca la deformación de los glóbulos rojos en "media luna" (célula falciforme) y desencadena una serie de trastornos sanguíneos que originan oclusión vascular con infarto de tejidos y órganos, principalmente del bazo (1-5).
En la costa peruana vive una importante población de raza negra y mestiza con ancestro africano, 10% y 2% respectivamente portan la hemoglobina S (Hb S) de manera asintomática (6-7). Además el índice de visitantes extranjeros a nuestro país ha crecido en los últimos años; por lo tanto, el riesgo de que sujetos con Hb S -no solamente con ancestro africano, sino provenientes del mediterráneo, medio oriente o la India- se expongan a la altura se ha incrementado.
Esta revisión tiene el propósito de transmitir los principales aspectos de esta entidad, para tenerla presente en el diagnóstico diferencial y tomar decisiones terapéuticas rápidas y apropiadas. Hay que reiterar que este desorden se agrava en pocas horas y pone en riesgo la vida del paciente.
Considero primordial impulsar medidas preventivas para que los individuos con ancestro africano o mediterráneo conozcan su estatus sickle cell antes de viajar a lugares por encima de los 2.500 m.
ASPECTOS MOLECULARES DE LA HEMOGLOBINA
El glóbulo rojo transporta el oxígeno -a través de la sangre-gracias a las moléculas de Hb que contiene. El glóbulo rojo tiene entre 250 a 300 millones de moléculas de Hb. (8).
La Hb humana está constituida por una molécula de globina a la cual se unen cuatro grupos HEM, generando un tetrámero que contiene los átomos de hierro. La globina tiene dos pares de cadenas de polipéptidos cada una de ellas contiene en promedio 145 aminoácidos (8-9)
La mayor parte de la población mundial posee HbA; esta Hb se considera normal. Un porcentaje importante de la población negra -de origen africano- posee una Hb diferente denominada Hb S, la cual conlleva numerosas consecuencias clínicas (9).
Normalmente un adulto tiene Hb A 97%, Hb A2 1-3% y Hb F 1%, esta última se encuentra en 100% durante la vida intrauterina y en 50-90% al momento del nacimiento.
GENÉTICA DE LA HEMOGLOBINOPATÍA S
La hemoglobinopatía es la enfermedad producida por la alteración de la molécula de Hb. La sustitución de un solo aminoácido de cualquiera de las cadenas peptídicas de la globina origina alteraciones físicoquimicas con implicancias clínicas y patológicas (8-10).
La hemoglobinopatía S es el resultado de una mutación en el sexto codón del gen beta-S (cromosoma 11) que sustituye adenina por timina. Este intercambio de nucleótidos se expresa con el correspondiente desplazamiento de un aminoácido (valina desplaza al ácido glutámico) en la posición 6 de la cadena beta de globina (8-11).
La hemoglobinopatía S es un desorden hereditario recesivo; sin embargo la Hb anormal es transmitida como dominante. La herencia de la Hb S sigue las reglas de la genética mendeliana clásica (9).
Cada individuo tiene dos genes para las cadenas beta de cada uno de los padres; si un individuo hereda un gen de una cadena beta anormal es un heterocigoto AS; si hereda los dos genes es un homocigoto SS.
Por consiguiente podemos calcular las posibilidades estadísticas cuando los dos padres son heterocigotos AS: 25% normales, 25% homocigotos y 50% heterocigotos. Si solamente uno de los padres es heterocigoto AS y el otro normal AA: 50% son normales y la otra mitad son heterocigotos.
FENÓMENO FALCIFORME Y OCLUSIÓN VASCULAR
La HbS desoxigenada es menos soluble y se agrega en largos polímeros (nucleación), estos polímeros se alinean entre si (polimerización) y forman una especie de gel insoluble que posteriormente cristaliza (10). La polimerización es el evento primario en la patogénesis molecular de la enfermedad sickle cell (11-16).
Los polímeros de HbS dañan la membrana del hematíe; también alteran las relaciones espaciales de las cadenas de globina (alfa y beta), distorsionando al glóbulo rojo en forma de media luna o de hoz (falciforme) (10). El fenómeno falciforme se incrementa con la deshidratación, acidosis, aumento de 2,3 difosfoglicerato (2,3-DPG) y los cambios de temperatura (1).
El hematíe falciforme o drepanocito es rígido y frágil, tiende a cohesionarse con otros glóbulos rojos, esta particularidad varía las propiedades reológicas de la sangre: aumenta la viscosidad y altera el flujo vascular (11).
Los hematíes falciformes -solos o agregados- no se adaptan a la red microvascular, tienen dificultades para deslizarse a través de la microcirculación y producen estásis. La estasis provoca vasoconstricción vascular, por consiguiente acrecentan la hipoxia y la deformación falciforme (14-16).
Los drepanocitos estancados pierden potasio y agua, además tienden a adherirse al endotelio vascular a través de diversas moléculas que se expresan en su superfi cie.
La cohesión de los drepanocitos y el consiguiente aumento de la viscosidad y estasis sanguínea, junto a la adherencia endotelial, van a conducir hacia la oclusión vascular con el consecuente daño irreversible de los tejidos (16).
La proteómica está identificando en la membrana y el citoplasma del glóbulo rojo falciforme la expresión de diversos grupos de proteinas, que permitirán entender mejor su compleja fisiopatología. Se han detectado defectos del citoesqueleto, disregulación de proteinas antioxidantes, disregulación de apolipoproteinas e incremento de proteinas de recambio o reparadoras (17-18)
DISTRIBUCIÓN GEOGRÁFICA DE LA HEMOGLOBINOPATÍA S
El gen de la Hb S es frecuente en el Africa central, especialmente en las regiones donde la malaria es endémica; está ampliamente distribuida alrededor de la línea ecuatorial de océano a océano (8-9).
Si bien la Hb S esta casi confinada a la población negra, el gen S también se encuentra en algunos países mediterráneos (Grecia, Italia, Israel), Arabia Saudita y la India (8). En Grecia uno de cada cinco pobladores de la región cercana al lago Kopais es portador de la Hb S (9); estudios de prevalencia de Hb S realizados en Kopais y Petromagoula encontraron 26.9% y 20.6% respectivamente.
El 8% de la población negra norteamericana y 7-13% de las poblaciones negras de América (Puerto Rico, Cuba, Panama, Colombia, Brasil, Venezuela, Jamaica, Curacao, Guadalupe y Martinique) tienen Hb S (8). No se la ha detectado en los aborígenes de Brasil, Bolivia y Perú.
La Hb S no se encuentra en la población aymara de Bolivia; sin embargo, Vázquez, Ergueta y Galarza (19) la han detectado en 6.45% de la población negra de los Yungas (valle cálido de poca altura en Bolivia).
En la población negra de la costa peruana Aste (6, 20) encontró 10% de Hb S y 7% de Hb C, Krumdieck (20) reportó 11.3% de Hb S y 3.5% de Hb C. Casas (21) y Jerí (22) estudiaron a 80 adultos y 20 niños de raza negra en dos hospitales de Lima y encontraron tres casos de Hb S y uno de Hb C.
El hallazgo de Hb C en los negros peruanos se debe a que sus ancestros provienen de la costa occidental del África, en donde la incidencia de Hb C es 12 al 15% (6).
Existen estudios de prevalencia de HbS en mestizos peruanos con ancestro africano: Hidalgo (7) en Talara encontró 1.3%, Tataje (23) en Lima 3%; Zegarra Villar (24) en Lima 4% y Casas (21) 2%.
Krumdieck (25) estudió a 87 mestizos peruanos (sin ancestro africano) y no encontró la Hb S. Roa y Col. (26) no ha encontrado hemoglobinopatías en recién nacidos mestizos de cerro de Pasco, Cusco y Puno.
Castillo (27) en 5.206 muestras de sangre estudiadas entre 1974 y 1996 en el hospital Rebagliati (Lima) han encontrado 218 (4%) con hemoglobina S (A/S 171, S/S 22, S/ beta-talasemia 20, S/C 4, S/O-Arabia 1 y 22 (0.42%) con hemoglobina C.
En el Perú existen reportes de hallazgos de hemoglobinopatía S en sujetos de raza blanca: Delgado (20) la detectó en un niño blanco, Jeri (22) la encontró en un bebe blanco en Talara y Casas la ha observado en un sujeto blanco de Caja-marca. Aste (6) y Frisancho (3) han observado sujetos blancos AS con infarto de bazo en la altura.
EXPRESIÓN CLÍNICA DE LA HEMOGLOBINOPATÍA S
Desde el punto de vista clínico existen tres grupos de pacientes con hemoglobinopatia S: portador AS, sickle cell homocigoto y sickle cell con doble hemoglobinopatía (1, 8-9).
A) Portador AS.- Es el sujeto heterocigotos AS, generalmente asintomático. No hay evidencia que tengan menos expectativa de vida, tampoco hay alguna limitación en la actividad habitual de los trabajadores manuales (8).
Pueden presentar algunas anormalidades como: hipostenuria, hematuria, necrosis papilar renal y bacteriuria asintomática en mujeres (9); asimismo infarto de bazo cuando se exponen a prolongada hipoxia (28-30), por ejemplo al ascender a grandes alturas.
Cuando se realizan ejercicios intensos la saturación de la sangre venosa tanto en normales como en portadores AS disminuye alrededor del 50%; in vitro el test de sicklemia ocurre cuando la saturación desciende al 25%, esta situación aparentemente brinda un margen de seguridad; sin embargo puede ser riesgosa si simultáneamente se presenta deshidratación, acidosis o cambios bruscos de la temperatura corporal (31-32).
Existen reportes de muerte súbita de atletas y reclutas AS sometidos a intenso entrenamiento físico (33-37). Se ha reportado rabdomiolisis, insuficiencia renal aguda y coagulación intravascular diseminada en soldados negros AS sometidos a intenso entrenamiento físico a mediana altitud (Colorado 2195 m) (34)
B) Sickle cell homocigoto.- Se refiere a los sujetos SS que poseen alto porcentaje de HbS (80-100%). Ellos presentan crisis de anemia hemolítica y fenómenos de oclusión vascular. La elevada viscosidad de las células deformadas puede culminar en infarto de varios órganos. Los niños tienen alto índice de morbimortalidad porque son vulnerables a las crisis vasooclusivas y hemolíticas (12).
C) Sickle cell asociado con otra hemoglobinopatia.- Se ha descrito que las personas con doble hemoglobinopatía (SC, SD y S-thalasemia) suelen tener manifestaciones clínicas mas severas (1, 15, 18).
HEMOGLOBINA S, INFARTO ESPLÉNICO Y ALTURA
En la década del cincuenta se comenzaron a reportar casos de infarto de bazo en sujetos con hemoglobinopatía S cuando volaban en aviones con cabina no presurizada (38-45).
En 1950 Sullivan (45) describió infarto de bazo en un soldado negro de 18 años que viajaba en un avión con cabina no presurizada; este paciente fue catalogado como "portador" AS, pero no se le realizó electroforesis.
En 1954 Cooley (46) reportó infarto masivo esplénico en seis soldados negros que se desplazaban -en aviones con cabina no presurizada- entre 3000 a 4600 metros de altura; a estos sujetos se les realizó esplenectomía; cuatro fueron catalogados como "portadores" AS y uno como SC.
Doenges (47) describió otros dos casos infarto de bazo en sujetos sickle cell en idénticas circunstancias que los pacientes anteriormente descritos.
En 1954, durante la guerra de Corea fueron descritos varios casos de infarto esplénico en soldados negros americanos aparentemente sanos, que volaban en aviones con cabina no presurizada; el cuadro se hacía clínicamente evidente entre 40 minutos a 2 horas de haber iniciado el viaje (40-42).
Coleman (48) ha descrito infarto esplénico durante el vuelo en sujetos AS y con doble hemoglobinopatía SC. Rotther (49) en 1956 describió seis casos más: tres AS, dos SC y uno S-Thalasemia.
Smith y colaboradores (50) en 1955 han descrito quince casos similares: once fueron AS, tres SC y uno S-thalasemia; a seis de estos sujetos se les practicó esplenectomía y se confirmó el diagnóstico de infarto de bazo.
Green (51) en 1971 ha descrito infarto de intestino delgado en una enfermera negra que se desplazó por via aérea; describió además seis SC y uno S-talasemia que presentaron infarto esplénico en viajes aéreos prolongados. Stock (52) ha descrito infarto esplénico en sujetos AS que volaban.
Actualmente viajar en un avión comercial con cabina presurizada equivale a volar entre 1.525 a 2.125 metros de altura, por consiguiente el riesgo potencial de los portadores AS es insignificante. El peligro de la aviación en sujetos homocigotos SS o con doble hemoglobinopatía es significante.
Se ha reportado infarto de bazo en sujetos expuestos a la hipoxia de altura (1-5). Obrien (53) ha descrito infarto de bazo en sujetos AS que estaban realizando alpinismo; Rywlin (54) describió un caso de infarto de bazo en un sujeto que cruzaba por vía terrestre los Andes en el Perú.
Criales (55) nos refirió que atendió a un deportista venezolano de 27 años con infarto de bazo, en los Juegos Bolivarianos realizados en La Paz-Bolivia en 1977. El diagnóstico precoz y la evacuación inmediata permitió su rápido restablecimiento (6); el estudio electroforético demostró que era portador AS heterocigoto.
Tapia-Gonzáles y Col (56). el año 2006 describieron que han atendido en el hospital de Mérida (Venezuela) a 11 pacientes con ancestro africano y hemoglobinopatía S (9 AS y 2 SS) que presentaron infarto de bazo cuando viajaban a Sierra Nevada (3800m) o se desplazaban en teleférico a Pico Espejo (4.765m). Los once pacientes fueron esplenectomizados de urgencia por presentar infarto masivo de bazo.
Morishima (57) y Funakoshi (58) el 2008 y 2010 respectivamente han visto a dos sujetos AS que presentaron infarto de bazo mientras escalaban el monte Fuji en el Japón.
INFARTO DE BAZO Y ALTURA EN EL PERÚ
Aste y Zavaleta (6, 59) dieron a conocer en 1959 el primer caso de infarto de bazo relacionado con la altura en el Perú. El paciente era un sujeto blanco de ascendencia belga y peruana, que se dirigía de Lima a Morococha (4,200 m); el estudio electroforético demostró que era un portador AS; posteriormente en Miami (USA) le realizaron esplenectomíia electiva, el estudio histopatológico demostró signos de infarto.
Aste (6) en 1961 describió dos casos más, se trataban de un cirujano del hospital Loayza y de un cadete de la Escuela Naval que se dirigían a La Oroya (vía terrestre) y a La Paz–Bolivia (vía aérea) respectivamente. Inicialmente aparentaron tener un cuadro de abdomen agudo quirúrgico; sin embargo, al ser evacuados a Lima mejoraron y no fue necesaria la cirugía.
Casas (21) en 1971 reportó un caso de infarto de bazo; en una mujer limeña AS de 36 años. El estudio familiar detectó la Hb S en su madre, hermana y uno de sus hijos.
En 1976 reportamos (60) el caso de un paciente AS con ascendencia árabe, que presentó infarto de bazo luego de trasladarse por vía aérea a Puno. El paciente fue intervenido quirúrgicamente debido a la ruptura espontánea del bazo; evacuado a Lima culminó su post-operatorio en buenas condiciones. La ruptura esplénica se debió a la demora diagnóstica.
Vega y Rivas (61) en 1975 han reportado dos casos fatales: uno de los pacientes fue un joven mestizo de 28 años que se trasladó de Piura al Cusco, donde presentó dolor abdominal. Fue intervenido quirúrgicamente sin detectar lesiones, lo que obligó al cirujano a cerrar el abdomen; posteriormente fue derivado a Lima donde fue reintervenido por múltiples perforaciones del ileón terminal y colon; el paciente falleció dos días después.
El intestino resecado tenía múltiples trombosis vasculares por hematíes falciformes. En este paciente el test de sicklemia fue positivo, pero no se realizó el estudio de electroforesis.
El otro caso se presentó en una niña negra de 8 años de edad, natural de Ica, quien viajó de Lima a Huancayo (3,200 m) presentando dolor abdominal. En el acto operatorio solo encontraron esplenomegalia, por lo que se cerró la cavidad abdominal. El post operatorio en Huancayo fue malo, por lo fue derivada a Lima donde falleció.
En la necropsia se encontró el bazo aumentado de volumen, intensamente congestionado, con zonas blanquecinas que correspondían a senos esplénicos trombosados por hematíes falciformes, los intestinos mostraron hallazgos similares. No se pudo precisar con exactitud si se trataba de un portador AS ó con doble hemoglobinopatia, por que no se realizó el estudio electroforético.
En 1977 un joven médico de 26 años, natural y procedente de Arequipa (2,300 m) viajó a Puno por ferrocarril para cumplir con su Servicio Civil de Graduandos; en el trayecto del viaje presentó dolor abdominal. Intervenido quirúrgicamente se encontró infarto esplénico y se realizó esplenectomía; fue evacuado a Arequipa, donde completó su recuperación (3).
En 1978 observamos dos casos más de individuos que presentaron infarto de bazo al dirigirse a Puno (26); uno de ellos fue un varón negro de 35 años de edad, natural de Chincha; y el otro, un estudiante universitario, mulato, de 21 años, natural de Casma y procedente del Callao (3).
En ambos casos se sospechó inicialmente de abdomen agudo, sin embargo los datos raciales, la esplenomegalia y el test de sicklemia positivo orientaron el diagnóstico. Las medidas de \oxigenación continúa y posterior derivación a la costa los recuperaron rápidamente. La electroforesis demostró que eran portadores AS.
En 1982 he tenido la oportunidad de observar en el hospital Rebagliati de Lima a un individuo AS blanco de 21 años que presentó dolor abdominal cuando se dirigía a las ruinas de Marcahuasi (sierra central); fue evacuado tres días después con un abdomen agudo. Intervenido quirúrgicamente se encontró un infarto masivo de bazo, se le realizó esplenectomía (3).
Castillo y Col. (27) refieren que en el Hospital Rebagliati (Lima) han observado entre 1974 a 1996 a cuatro pacientes AS que presentaron infarto esplénico cuando se desplazaban a regiones altas; uno de ellos presentó también infarto pulmonar.
Chacaltana y Col (62) en el 2004 reportó a un paciente varón AS que presentó infarto de bazo al llegar a Obrajillo (2.764 m), una localidad de la sierra de Lima, fue evacuado a Lima, donde llegó a las 30 horas de iniciado el proceso; el paciente evolucionó bién.
En el 2006 Ruiz y Col. (63) observaron en el Hospital Loayza (Lima) a un paciente AS varón mestizo de 21 años con infarto de bazo, derivado de la ciudad minera de Casapalca (4200 m); su rápida transferencia a Lima permitió su recuperación sin complicaciones.
En el 2009 López de Guimaraes y Col. (64) reportaron tres casos de turistas heterocigotos AS con infarto de bazo en Huaraz (3100 m): El primero fue un cubano blanco de 55 años que fue esplenectomizado, el segundo fue un cubano negro de 23 años, y el tercero fue un limeño mestizo de 17 años; los dos últimos pacientes fueron diagnosticados precozmente y derivados a Lima sin complicaciones.
CARACTERÍSTICAS CLÍNICAS DEL INFARTO DE BAZO EN LA ALTURA
Edad y género.- Los pacientes son jóvenes (51-61), las edades fluctuan entre 18 y 34 años, con un promedio de 25 años, La mayoría fueron del género masculino. Estos hallazgos están relacionados con el mayor desplazamiento geográfi co de los hombres jóvenes.
Raza.- La mayoría son negros y mestizos con ascendencia africana; existen reportes de sujetos blancos con ascendencia mediterránea (65-69). He visto a un estudiante brasileño blanco (con ancestro mediterráneo) con infarto de bazo en Puno (3).
Procedencia.- Todos los pacientes se desplazaban por primera vez a regiones de altura; se dirigieron de la costa a la sierra en transporte terrestre o aéreo (3, 6, 62-64). La mayoría vivían en la costa peruana¸ también se han visto pacientes procedentes de Brasil, Ecuador, Venezuela, entre otros.
En nuestra serie (3) distribuimos a los pacientes por su origen de nacimiento: 3 de Lima, 1 del Callao, 1 de Casma, 1 de Chincha, 1 de Cañete, 1 de Arequipa, 1 de Curitiba (Brasil) y 1 de Guayaquil (Ecuador).
Cuadro clínico.- El cuadro clínico se caracteriza por ser de inicio agudo y de carácter progresivo; aparece en el trayecto del viaje, generalmente cuando se han pasado los 3,000 metros de altura (51-70).
Los pacientes presentan dolor abdominal, generalmente a predominio de hipocondrio izquierdo; a veces difuso o referido hacia hemitórax izquierdo. La intensidad aumenta progresivamente.
El examen abdominal inicialmente puede ser normal, conforme pasan las horas se puede detectar esplenomegalia. La demora del diagnóstico influye en la aparición de signos de irritación peritoneal. En un paciente se detectó abdomen agudo con signos de anemia aguda, por hemorragia interna consecutiva a la ruptura espontánea del bazo (60).
La presentación de fiebre, naúsea, vómitos y de molestias generales (malestar general, astenia o decaimiento) es variable de paciente a paciente; depende también del tiempo de enfermedad transcurrido.
No es inusual, que las molestias generales del cuadro falciforme se superpongan con las del Mal de Montaña Agudo (Soroche Agudo); situación, que contribuye a confundir el diagnóstico principal (3).
Una buena anamnesis y un buen examen físico nos pueden dar las bases para plantear el diagnóstico de infarto de bazo asociado a hemoglobinopatía S. La sospecha clínica es crucial en la etapa inicial, para evitar complicaciones (3).
Si desconocemos esta entidad, no la vamos a plantear en el diagnóstico diferencial, por consiguiente nuestras decisiones van a ser erróneas, y ponemos en peligro la vida del paciente.
Exámenes auxiliares.- Se deben solicitar los exámenes auxiliares que habitualmente se utilizan para evaluar un abdomen agudo quirúrgico: hemograma, orina, bioquímica, perfil de coagulación, entre otros.
Los pacientes habitualmente presentan leucocitosis, con aumento de sus formas no maduras ("desviación izquierda"), este dato hay que tenerlo presente, para no tomar decisiones erróneas o precipitadas.
El test de Sicklemia o prueba del Falciformismo es útil; la prueba se realiza mezclando una gota de sangre con un agente reductor como el bisulfito de sodio al 2%, minutos después se pueden observar a los glóbulos rojos deformados, la mayoría en forma de media luna (3, 7).
La electroforesis del hemolizado del glóbulo rojo es mas específica; en este examen las hemoglobinas migran en un campo eléctrico de acuerdo a la magnitud de su carga, lo que permite identificarlas.
Las hemoglobinas con similar comportamiento electroforético las podemos diferenciar mediante exámenes como cromatografía, prueba de solubilidad, prueba de la inestabilidad de la oxihemoglobina, entre otros.
La sospecha clínica y el test de sicklemia rápidamente pueden orientar el diagnóstico para instaurar un tratamiento precoz: oxigenación adecuada y traslado inmediato a un medio de menor altitud (3).
Estudios de imágenes.- La radiografía de abdomen simple en ocasiones nos permite reconocer el crecimiento esplénico que eleva el hemidiafragma izquierdo o rechaza la vísceras (estómago o intestinos) hacia la derecha del abdomen; éste estudio también nos sirve para apreciar el compromiso abdominal (ileo paralítico, etc.).
La radiografía de tórax puede mostrarnos derrame pleural izquierdo, asociado a la extensión transdiafragmática del infarto esplénico o lesiones concurrentes de infarto pulmonar.
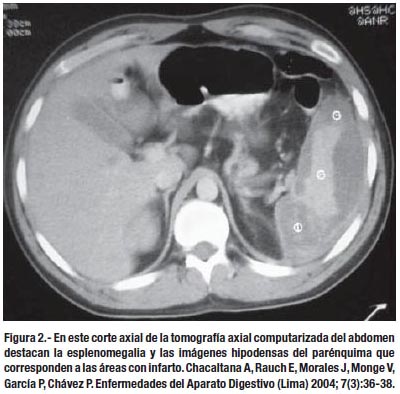
La ecografía abdominal y la tomografía axial computarizada de abdomen nos permiten observar el interior del bazo, destacando en el parénquima las imágenes -hipoecogénicas o hipodensas respectivamente- de las áreas de infarto (62).
En la tomografía dinámica las lesiones en forma "de cuña" con la base amplia dirigida hacia la cápsula y el ápex dirigido hacia el hilio son sugestivas de infarto; asimismo, el contraste puede acentuar la periferie esplénica por la mayor perfusión de los vasos capsulares. Si hay extensa necrosis por licuefacción, la lesión hipodensa puede mostrar nivel aire-fluido o burbujas.
La ecografía doppler permite reconocer la ausencia de flujo en las zonas de infarto; el angiograma también demuestra las zonas de hipoperfusión esplénica.
MEDIDAS TERAPÉUTICAS
El diagnostico precoz es fundamental, permite la instauración rápida de medidas como reposo absoluto, hidratación parenteral y oxigenación continua adecuada hasta su evacuación a localidades de menor altitud (3). Con estas medidas oportunas se atenua el fenómeno del falciformismo y los pacientes pueden superar este trance agudo sin presentar complicaciones (64, 70).
El retardo diagnótico conlleva a tomar medidas que pueden incluso exacerbar la hipoxia tisular y producir infarto masivo de bazo (60) o de otros órganos.
Si actuamos adecuadamente dentro de las primeras horas y evacuamos al paciente el primer día, alejamos la posibilidad de presentación de complicaciones mayores (3,70).
La intervención quirúrgica tiene alto riesgo, y se debe practicar en situaciones dramáticas como la presentación de un infarto masivo o la ruptura del bazo (60) para evitar las consecuencias de la peritonitis o la hemorragia interna.
HALLAZGOS ANÁTOMOPATOLOGICOS
El bazo por su tipo circulatorio -falta de comunicaciones laterales entre las ramas de la arteria esplénica- es un órgano muy susceptible de la crisis falciforme (3,15).
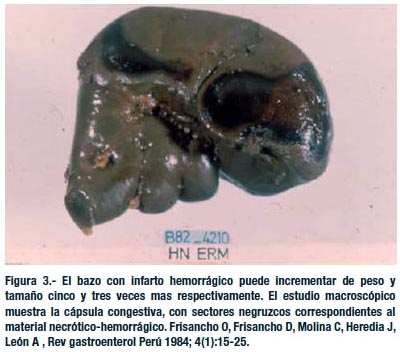
El bazo normalmente -en promedio- pesa 150 gramos y mide 12 x 7 x 3 cm. El bazo cin infarto hemorrágico puede aumentar cinco veces su peso y tres veces su tamaño (1, 3, 61).
El estudio macroscópico del bazo infartado nos muestra la cápsula congestiva, con sectores negruzcos correspondientes al material necrótico-hemorrágico. Al corte el parénquima congestivo es friable, contiene bandas hemorrágicas alrededor de las zonas infartadas (3, 12, 14).
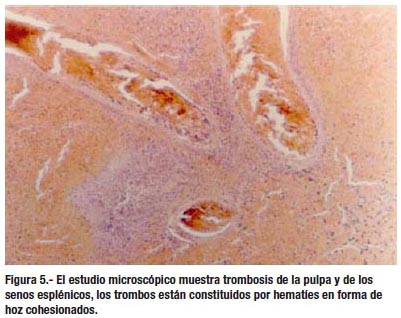
El estudio microscópico muestra trombosis de la pulpa y de los senos esplénicos, los trombos están constituidos por hematíes en forma de hoz cohesionados (3, 61). El margen del infarto está separado del parénquima residual por tejido con abundante infi ltrado inflamatorio (61).
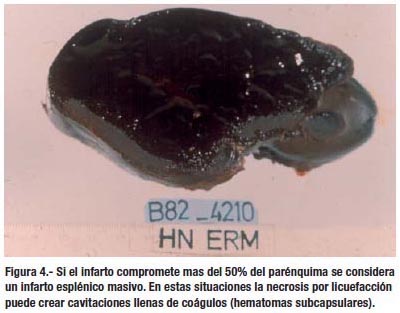
Si el infarto compromete mas del 50% del parénquima se considera un infarto esplénico masivo. En estas situaciones la necrosis por licuefacción puede crear cavitaciones llenas de coágulos (hematomas subcapsulares) (3).
El infarto esplénico en la altura -en correspondencia a diferentes circunstancias- puede evolucionar en tres etapas (3): a) Infarto agudo (focal, no complicado), b) infarto masivo y c) infarto con disrupción capsular (rotura espontánea) (60).
Conjuntamente con el infarto de bazo se ha reportado infarto en otros órganos (intestino, hígado y pulmones); es posible que en algunos casos estén presentes de manera subclínica, encubiertos por la intensa expresividad del daño esplénico.
FACTORES RELACIONADOS CON LA SEVERIDAD CLÍNICA
Reiteramos que la demora diagnóstica y el retardo en la evacuación del paciente van a provocar que las manifestaciones clínicas del infarto esplénico se agrave conforme pasan las horas (3, 60).
Otros factores que están vinculados con la severidad falciforme son: el grado de hipoxemia, porcentaje de Hb S, haplotipos de HB S, Hb S con doble mutacion, nivel de Hb fetal, portador de doble hemoglobinopatía y défi cit de G6PDH; también influyen el estado de hidratación, acidosis, estrés oxidativo, nivel de 2,3 DPG y los cambios bruscos de temperatura (71-79).
Existen algunos reportes sobre sujetos AS que toleran bién medianas alturas. Green (51) refirió que los atletas africanos AS que compitieron en las Olimpiadas de Mexico 1968 no presentaron ninguna manifestación clínica asociada a crisis drepanocítica.
El 8% de los deportistas negros de la liga de fútbol americano (USA) son portadores AS; la mayoría de ellos ha participado en competencias a medianas alturas -como en Colorado (2,100 m.)- sin presentar problemas (36).
Nosotros hemor reportado (3) el caso de un sujeto AS que vivía normalmente en Arequipa (2.300 m) y que presentó infarto esplénico al ascender a Puno (3.850m); y es que la curva de disociación de la oxihemoglobina nos muestra que encima de los 2500 mt de altura, ascensos limitados de altitud (pequeñas reducciones de la presión barométrica) pueden provocar caidas enormes de la saturación de oxígeno arterial (6)
Los reportes antes mencionados indican que la presión parcial de oxígeno a medianas alturas no es crítica para deformar los glóbulos rojos en porcentajes clínicamente peligrosos; sin embargo por encima de los 2500 m la afinidad de la hemoglobina S por el oxígeno disminuye notablemente.
El porcentaje de hemoglobina S que se lleva en la sangre es el principal factor relacionado con la expresión clinica (49); si es alta, es menor la posibilidad de tolerar pequeñas caídas de la saturación de oxígeno. Además en el laboratorio se ha demostrado que en condiciones de hipoxia existe una correlación positiva entre la concentración de Hb S y su polimerización.
Rother(49) observó que el porcentaje de Hb S (42-44%) de los pacientes AS con infarto de bazo fue alto, comparado con el de otros sujetos AS que tuvieron niveles menores de 40%.
Con el desarrollo de nuevas tecnologías se han identifi cado seis haplotipos del gen beta-S (72-73). El haplotipo CAR o Bantú expresa una enfermedad agresiva, el haplotipo Senegal se asocia a cuadros benignos, el haplotipo Benin a cuadros intermedios; en otras palabras se están reconociendo varios subtipos de Hb S con manifestaciones clínicas diferentes.
La Hb S Antillas es una Hb con doble mutación (alfa2 y beta2) que está asociada con falciformismo sintomático en estado heterocigoto (72). Esta variante tiene la misma movilidad electroforética de la Hb S. Los glóbulos rojos con Hb S Antillas inician la malformación falciforme (sickling) con el mismo nivel de presión parcial de oxígeno que la inducen en los hematíes SC. Otra Hb S con doble mutación en el alelo beta recientemente descubierta es la Hb S-Wake beta (73).
El efecto de otra hemoglobina anormal concurrente (doble hemoglobinopatía) empeora el cuadro clínico (74-76). Se ha descrito drepanocitosis y sus complicaciones en sujetos con SC, SD y S-talasemia (beta, Lepore, HPFH).
Githen (76) ha descrito el síndrome de secuestración esplénica en personas con Hb S expuestas a la altura, el cual se caracteriza por el inicio súbito de dolor en el hipocondrio izquierdo y esplenomegalia. Entre los principales hallazgos destacan: anemia, trombocitopenia y scans con el bazo hipofuncionante. En este reporte se menciona que los sujetos con doble hemoglobinopatía SC tienen mas riesgo de presentar secuestración esplénica que los portadores AS.
Konotey (1,15) cuestiona algunos de los reportes iniciales de infarto de bazo en sujetos catalogados como "portadores AS", porque no tenían estudios de electróforesis de hemoglobina, indicando que algunos casos podrían corresponder a desórdenes con doble hemoglobinopatía.
El grado de desoxigenación requerida para producir deformidad de los glóbulos rojos en un AS es mayor que la requerida para un homocigoto SS; los glóbulos rojos de los sujetos con doble hemoglobinopatía SC o S-thalasemia se sitúan en un rango intermedio (8).
Ha sido bien estudiada la interacción de otras hemoglobinas anormales, algunas de las cuales tienen gran habilidad para copolimerizar con la hemoglobina S acentuando los fenómenos sickling: la hemoglobina C tiene esta característica (10-11).
La hemoglobina F (fetal) no participa en la formación del polímero, al contrario inhibe el fenómeno sickling (77). Los recién nacidos homocigotos SS retardan su expresión clínica hasta cuando disminuyen sus niveles de Hb F (9).
Existen sujetos homocigotos SS con altos niveles de Hb fetal -porque portan el gen de la persistencia de la Hb F- que solo presentan manifestaciones clínicas si el nivel de Hb F es menor de 35%; si la Hb F supera el 10% el curso de la enfermedad es moderado. Desafortunadamente la mayoría de los sujetos SS habitualmente solo tienen 5% de hemoglobina F.
Los sujetos Hb S en Arabia Saudita y en las tribus Veditas de la India (77) presentan cuadros clínicos benignos, porque portan -la mayoría de ellos-niveles de Hb F de 15 a 30 %. Esta situación hay que tenerla en cuenta, cuando estudiemos el amplio rango de severidad de la hemoglobinopatía S en los diferentes grupos étnicos.
Otro factor que exacerba el fenómeno del falciformismo es la deficiencia de glucosa 6 fosfato deshidrogenasa (G6PDH), Konotey (15) reportó que la falta de esta enzima empeoró el cuadro clinico falciforme de sus pacientes.
La enzima G6PDH participa del metabolismo oxidativo del glóbulo rojo. La carencia de esta enzima disminuye la capacidad del hematíe para enfrentar agresiones oxidantes y facilita su atrapamiento y destrucción (hemólisis) en el RES del bazo (78-79).
La prevalencia del déficit de la G6PDH en afroamericanos (USA) es 12%, bantúes africanos 20%, negros brasileros 8% y blancos mediterráneos 35%; en el Perú la prevalencia es de 5% en negros (Chincha) y 0.71% en mestizos peruanos (78).
Podemos concluir que el comportamiento clínico de los sujetos con hemoglobinopatía S difiere, porque los aspectos fisiopatológicos son mas complejos y dependen de la concurrencia de varios factores.
REFERENCIAS
1. KONOTEY AHULU FID. Sickle cell trait and altitude. Br Med J 1972; 1: 177.
2. CLASTER S, GODWIN MJ, EMBURY SH. Risk of altitude exposure in sickle cell disease. West J Med 1981; 135(5):364-367.
3. FRISANCHO O, FRISANCHO D, MOLINA C, HEREDIA J, LEÓN A. Infarto de bazo y altura, Rev Gastroenterol Perú 1984; 4(1):15-25.
4. NUSSBAUM RL, RICE L. Morbidity of sickle cell trait at high altitude. South Med J 1984; 77(8):1049-1050.
5. GOLDBERG NM, DORMAN JP, RILEY CA, ARMBRUSTER EJ Jr. Altitude related specific infarction in sickle cell trait case reports of a father and son. West J Med 1985; 143(5):670-672.
6. ASTE SALAZAR R. Contribución peruana al estudio de la biología de las grandes alturas. Revista del Viernes Médico 1974; 1: 24-25.
7. HIDALGO E. Anemia de células falciformes en la ciudad de Talara. Tribuna Médica del Perú 1966; 11: 89-90.
8. SERJEANT GR, BERYL AS. Sickle Cell Disease. 3rd ed. New York: Oxford University Press; 2001
9. STUART MJ, NAGEL RL. Sickle-cell disease. Lancet 2004; 364:1343-1360.
10. SAMUEL RE, SALMON ED, BRIETL RW. Nucleation and growth of fibres and gel formation in sickle-cell haemoglobin. Nature 1990; 345:833-855.
11. GOODMAN SR. The irreversibly sickled cell: a perspective. Cell Mol Biol 2004; 50(1):53-58.
12. TSARAS G, OWUSU-ANSAH A, BOATENG FO, AMOATENG-ADJEPONG Y. Complications associated with sickle cell trait: a brief narrative review. Am J Med 2009; 122(6):507-512.
13. MOTULSKY AG. Sicklemia. JAMA 1954; 155: 388-389.
14. DIGGS LW. Anatomic lesions in sickle cell diseases. Sickle cell Disease. Abramson-Bertles-Wethérs C.V. Mosby, St. Louis 1973.
15. KONOTEY AHULU FID. The sickle cell disease: clinical manifestation including the "sickle crisis". Arch Inter Med 1974; 133: 611-619.
16. KAUZ DK, FABRY ME, NAGEL RL. The pathophysiology of vascular obstruction in the sickle syndroms
17. YUDITSKAYA S, SUFFREDINI AF, KATO GJ. The proteome of sickle cell disease: insights from exploratory proteomic profiling. Expert Rev Proteomics 2010, 7(6): 833-848.
18. KAKHNIASHVILI DG, GRICO NB, BULLA LA, GOODMAN SR. The proteomic of sickle cell disease; profiling of erythrocyte membrane proteins by 2D-DIGE and tandem mass spectrometría. Exp Biol Med 2005; 230(11):787-792.
19. VÁZQUEZ E, ERGUETA J, GALARZA M. Hemoglobina S en Bolivia. Tesis Facultad de Ciencias Farmacéuticas y Bioquímicas, Universidad Mayor de San Andrés, La Paz, Bolivia, 1971, 21 pag.
20. ASTE SALAZAR H. Diferenciación de hemoglobinas en la población negra de Lima. Anales de la Facultad de Medicina 1957; XL (4): 866-868.
21. CASAS LS. Hemoglobinopatias en el Hospital Central del Empleado (determinaciones electroforénicas en 100 pacientes de raza negra). Tesis Bachiller de Medicina Nº 2585, UNMSM, Lima - Perú 1971.
22. JERÍ A. Investigación de hemoglobinas anormales mediante electroforesis en acetato de celulosa. Tribuna Médica del Perú 1972; 31 (9): 314-316.
23. TATAJE J. La anemia falciforme y la drepanocitosis en nuestro medio. Tesis Bachiller de Medicina, UNMSM, Lima Perú 1958.
24. ZEGARRA VILLAR G. Estudio clínico y hematológico de la anemia falciforme y del fenómeno llamado sicklemia. Tres casos interesantes. Tesis Bachiller de Medicina, UNMSM, Lima-Perú 1945.
25. KRUMDIECK BOIT CL. Anomalias hereditarias en la síntesis de la hemoglobina. Tesis de Bachiller de Medicina Nº 4482 UNMSM, Lima - Perú, 1958.
26. ROA D, AGUINAGA MP, RUIZ W, ULLOA V, TURNER E. Búsqueda de hemoglobinas anormales en los recién nacidos en las grandes alturas. Rev Med Hered 1997; 8(3): 87-91.
27. CASTILLO J, HAZÁN E, MÁRQUEZ MC. Las hemoglobinopatías en el Perú. Revista Médica del IPSS 1998; 7(1): 7-17.
28. CHAMBERLAND DL. Splenic infarction in an african american with sickle cell trait. Am J Hemat 2006; 82:86-87.
29. FRANKLIN QJ, COMPEGGIE M. Splenic syndrome in sickle cell trait: four case presentations and a review of the literature. Mil Med 1999; 164(3):230-233.
30. LEVIN EC, BAIRD WD, PERRY JE, et al. The experimental production of splenic sequestration of erythrocytes in patient with sickle cell trait. J Lab Clin Med 1957; 50: 926-930.
31. CONNES P, HARDY-DESSOURCES MD, HUE O. Counterpoint: Sickle-cell trait should not be considered asymptomatic and as a benign condition during physical activity. J Appl Physiol 2007; 103(6):21382140.
32. YORK L, BRIERRE JI. How diligently should the diagnosis of sickle cell trait be pursued clinical case report with military implication. Milit Med 1971; 136: 27-29.
33. KARK JÁ, POSEY DM, SCHUMACHER HR, RUEHLE CJ. Sickle-cell trait as a risk factor for sudden death in physical training. N Eng J Med 1987; 317:781-787.
34. KOPPES GM, DAL Y JJ, COLTMAN CA, BUTKUS DE. Exertion induced rhabdomyolisis with acute renal failure and disseminated intravascular coagulation in sickle cell trait. Am J Med 1977; 63:313-317.
35. GRADNER JW, KARK JÁ. Fatal rhandomyolisis presenting as mild heat illness in military training. Mil Med 1994; 159(2):160-163.
36. MURPHY JR. Sickle hemoglobin (AS) in black football players, JAMA 1973; 225:981-982.
37. JONES SR, BINDER RA, DONOWHO EM Jr. Sudden death in sickle-cell trait. N Eng J Med 1970; 282: 323-325.
38. CANBY CB, CARPENTER G, ELLMORE LF. Drepanocytosis (sicklemia) and an apparently acute surgical condition of the abdomen. Arch Surg 1944; 48: 123-125.
39. HENDERSON AE, THORNELL HE. Observations on affecting lowered oxygen tension in sicklemia and sickle cell anemia in army fIying personnal. J Lab Clin Med 1946; 31: 769-771.
40. CONN HO. Sickle cell trait and splenic infarction associated with high altitud e fIying. N Eng J Med 1954; 251: 417-419.
41. MOTULSKY AG, LUTTGENS WF, PETTERSON W, ROTTER R. Splenic infarction precipitated by airplane flight in patients with sicklemia Clin Resp Proc 1955 3: 51-55
42. FINDLAY GM, BOULTER EA, MAC GIBBON CE. A note sickling and fIying. J. Roy Army Med Corps 1957; 89: 138-140.
43. HENRY CM Jr. Sickle cell crisis without anemia, ocurring during air flight. Milit Surg 1964;115: 271-274.
44. McKENZIE JM. Evaluation of the hazards of sickle cell trait in aviation. Aviat Space Environ Med 1977; 48: 753-755
45. SULLIVAN BH. Danger of airplane flight to persons with sicklemia. Ann Int Med 1950; 32: 338-340.
46. COOLEY JH, PETERSON AL, ENGELS CE, JERNIGAN JP. Clínical triad of massive splenic infarction, sicklemia trait and hight altitude flyng. J Am Med Assoc (JAMA) 1954; 154: 111-113.
47. DOENGES JP, SMITH EW, WISE SP, BREITENBUCHER RB: Splenic infarction following air travel and associated with the sickling phenomenon. JAMA 1955; 156: 114-117.
48. COLEMAN WA, FURTH FW. Splenic infarction in a patient with sickle cell hemoglobin C disease. Arch Inter Med 1956; 98: 247-249.
49. ROTTER R, LUTTGENS WF, PETERSON WL, STOCK AE, MOTULSKY AG. Splenic infarction in sicklemia during airplane flight. Pathogenesis, hemoglobin análisis and clínical features of six cases. Ann Int Med 1956; 44: 257-258.
50. SMITH EW and CONLEY CL. Sicklemia and infarction of the spleen during serial flight. Electroforesis of the hemoglobin in 15 cases. Bull Johns Hopkins Hosp 1955; 96:35-37.
51. GREEN RL, HUNTSMAN RG, SERJEANT GR. The sickle-cell and altitude. Br Med J 1971; 4(5787): 593-595.
52. STOCK AE. Splenic infarction associated with high altitude fIying and sickle cell trait. Ann Int Med 1956; 44: 554-556.
53. OBRIEN RT, PEARSON HD, GODLEY JA, SPENCER RP. Splenic infart and sickle-cell trait. N Eng J Med 1972; 287: 720-724.
54. RYWLIN AM, BENSON J. Massive necrosis of the spleen with formation of a pseudo cyst. Report of a case in a white man with sickle cell trait. Am J Clin Pathol 1961; 36: 142-144.
55. CRIALES H.. Drepanocitosis en la altura. XII Jornada Quirúrgica Nacional, Sociedad Boliviana de Cirugía. Cochabamba, agosto 1978.
56. TAPIA CONZÁLES JL, GONZÁLES G, SÁNCHEZ A, UZCÁTEGUI E, GUZMÁN JL, CAMARATA F. Infarto esplénico por anemia falciforme relacionado con la altura. Rev Venez Cir 2006; 59(2):60-65.
57. MORISHIMA A, SCHOFER JM, PELLETIER P, McKEE SM. Images in emergency medicine: splenic infarction due to sickle cell trait after climbing Mt. Fuji. West J Emerg Med 2008; 9(3):179.
58. FUNAKOSHI H, TAKADA T, MIYAHARA M, et al. Sickle cell trait as a cause of splenic infarction while climbing M. Fuji. Intern Med 2010; 49(16): 1827-1829.
59. ZAVALETA AF. El falciformismo de los hematies como causa de infarto esplénico en la altura (presentación de 3 casos). Tesis Bachiller de Medicina Nº 5530, UNMSM, Lima Perú 1962.
60. FRISANCHO D, DEL CASTILLO A, FRISANCHO O. Infarto y ruptura espontánea del bazo en la altura. Tribuna Médica del Perú 1977; XL(4):494-496.
61. VEGA L y RIVAS L. Drepanocitosis. Tribuna Médica del Perú 1975; 959:321-322.
62. CHACALTANA A, RAUCH E, MORALES J, MONGE V, GARCÍA P, CHÁVEZ P. Dolor abdominal agudo como manifestación de hemoglobinopatìa estructural heterocigota (rasgo falciforme o sickle trait). Enfermedades del Aparato Digestivo 2004; 7(3):36-38.
63. RUIZ E, GARAVITO J, JIMENEZ J, ARTEAGA R, GARCÌA JL, CHÁVEZ V. Dolor abdominal agudo debido a infarto esplênico en un paciente con enfermedad heterocigota de células falciformes expuesto a la altura. Rev Gastroenterol Perù 2006; 26:386-389.
64. LÓPEZ DE GUIMARAES D, MENACHO J, VILLANUEVA J, MOSQUERA V. Infarto esplénico en la altura, Huaraz-Perú (3.100m). Rev Gastroenterol Perú 2009;29:179-184.
65. DIEP BN, SCHEIRMAN K, REEVES WB, MASK DR, EICHNER ER. Splenic infarction in a white man with sickle cell trait. South Med J 1979; 72:1611-1613.
66. COX RE. Splenic infarct in a White man with sickle cell trait. Ann Emerg Med 1982; 11:668-669.
67. LANE PA, GITHENS JH. Splenic syndrome at mountain altitudes in sickle cell trait. Its occurrence in nonblack persons. J Am Med Assoc (JAMA) 1985; 253:2251-2254.
68. TIERNAN CJ. Splenic crisis at higt altitude in 2 white men sickle cell trait. Annals of Emergency Medicine 1999; 33(2):230-233
69. SHALEV O, BOYLEN AL, LEVENE C, OPPENHEIM A, RACHMILEWITZ EA. Sickle cell trait in a white jewish family presenting as splenic infarction at high altitude. Am J Hematol 1988; 27(1):46-48.
70. SHEIKA A. Splenic syndrome in patients at high altitude with unrecognized sickle cell trait: splenectomy is often unnecessary. Can J Surg 2006; 48(5):377-381.
71. SCHECHTER AN, BUNN HF. Whats determines severity in sickle-cell Disease. N Eng J Med 1982; 306: 295-297.
72. MONPLAISIR N, MERAULT G, POYART C, et al. Hemoglobin S Antilles: a variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozigotes. Proceedings of the National Academy of Sciences of the United States of America 1986; 83(24):9363-9367.
73. KUTLAR F, REDDING-LALLINGER R, MEILER SE, et al. A new sickling variant Hb S-wake beta found in a compound heterozygote with Hb S beta coinherited with homozygous alfa-talasemia-2: phenotype and molecular characteristics. Acta Haematol 2010; 124(2): 120-124.
74. HIGGS DR, ALDRIDGE RE et al. The interaction of alpha thalassemia and homozygous sickle cell disease. N Eng J Med 1982; 306:1441-1446.
75. USTUN C, KUTLAR F, HOLLEY L, et al. Interaction of sickle cell trait with hereditary spherocytosis. Splenic infarct and sequestration. Acta Haematol 2003; 109(1):46-49.
76. GITHENS JH, PHILLIPS CR, HUMBERT JR, BONNER RN, EWING PC. Effects of altitude in persons with sickle hemoglobinopathies. Rocky Mountain Medical Journal 1975; 515-519.
77. PERINE RP. Natural history of sickle cell anemia in Saudi Arabs. A study of 270 subjets. Ann Int Med 1978; 88: 1-6.
78. RUIZ W, ULLOA V, BAYLÓN O. Prevalencia de la deficiencia de G6PDH en donadores voluntarios de sangre que acuden a los hospitales Cayetano Heredia y Arzobispo Loayza. Lima-Perú. Rev Med Hered 1997; 8(1):11-18.
79. HELLER P, BEST WR, NELSON RB, BECKTEL J. Clinical implications of sickle-cell trait and glucose-6 phosphate dehydrogenase deficiency in hospitalized black male patients. N Engl J Med 1979; 3000:1001-1005.
