











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Recientemente y de acuerdo a la clasificación de la Organización Mundial de la Salud (OMS) de 2022 1, la denominación de adenomas hipofisiarios ha cambiado por el término tumores neuroendocrinos de la pituitaria (PitNET). Los tumores hipofisiarios típicamente crecen lentamente y puede ser controlados por cirugía y/o tratamientos farmacológicos, el crecimiento se da en 20% de los pacientes y tras 10 años de tratamiento quirúrgico, el 50% de los tumores recidiva 2.
Aunque la mayoría de ellos presentan un fenotipo benigno, un pequeño subgrupo exhibe una presentación intermedia con crecimiento local agresivo, con capacidad de invadir el seno esfenoidal, cavernoso, con crecimiento supraselar y recidivas, constituyendo un importante reto diagnóstico y terapéutico. Estos tumores pituitarios agresivos tienen una prevalencia desconocida son más frecuentes en varones, invaden el seno cavernoso en 80% y son gigantes en 22% 3.
La presencia de invasión y tendencia a recidivar hace altamente recomendable que estos tumores sean intervenidos por neurocirujanos de experiencia, y reintervenidos tantas veces como sea necesario para reducir los síntomas de compresión local. La radioterapia adyuvante no debería demorar cuando el crecimiento tumoral o hipersecreción hormonal no puedan controlarse con la cirugía. Todo eso hace importante el abordaje por un equipo multidisciplinario. En el presente caso de infrecuente presentación, corresponde a un paciente con acrogigantismo asociado a tirotoxicosis por macroadenoma hipofisiario invasivo funcionante. Para la publicación de este reporte se obtuvo el consentimiento informado de un familiar directo del paciente.
REPORTE DE CASO
Varón de 23 años, procedente de Venezuela, que desde los 16 años cursó con bocio sin presencia de síntomas compresivos asociado a temblor distal, diaforesis, hiperfagia y pérdida de peso de 20 kg en un año. Por lo descrito, acude a los servicios de salud donde le diagnostican hipertiroidismo central e inician tratamiento con tiamazol 15 mg/día y propranolol 80 mg/día.
A los 17 años cursó con incremento de la velocidad de crecimiento (altura 1,86 metros, altura genética esperada 1,73 m) y cefalea hemicraneana izquierda opresiva asociado a disminución de la agudeza visual izquierda que progresó a amaurosis. Le indicaron tratamiento quirúrgico que no llegó a realizarse, se comprobó hipersecreción de hormona de crecimiento por lo que se le agregó octreotide 20 mg mensual en forma irregular, no acudió a controles por tres años y emigró a Perú. A los 21 años, paciente ingresó por Emergencia del Hospital Nacional Dos de Mayo, con diagnóstico de tormenta tiroidea inminente. Se confirmó bioquímicamente exceso de producción de hormonas tiroideas, prolactina y de hormona de crecimiento (Tabla 1).
Tabla 1. Exámenes de laboratorio basal, posterior a exéresis parcial de macroadenoma hipofisiario y tiroidectomía total.
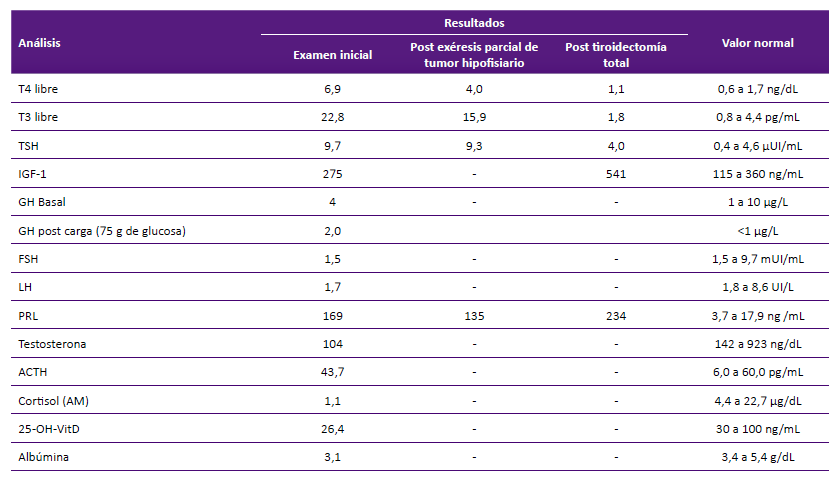
TSH: hormona estimulante de la tiroides, IGF-1: factor de crecimiento similar a la insulina 1, GH: hormona de crecimiento, FSH: hormona foliculo estimulante, LH: hormona luteinizante, PRL: prolactina, ACTH: hormona adrenocorticotropica
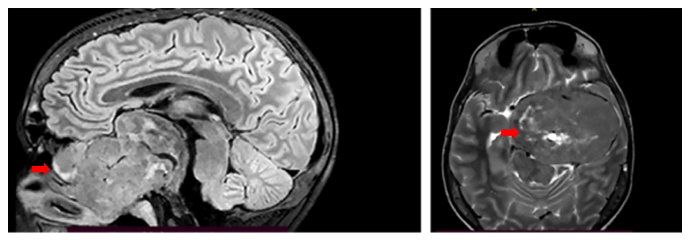
Se realizó una resonancia magnética de hipófisis encontrándose un tumor a nivel se-lar con extensión supraselar de 7 cm x 8 cm x 5 cm con compromiso de seno esfenoidal y ambos senos cavernosos, quiasma óptico, lóbulo temporal izquierdo y mesencéfalo (catalogado como Knosp 3b) (Figura 1).
El paciente fue transferido a neurocirugía para intervención quirúrgica de craneotomía descompresiva frontotemporal izquierda y exéresis parcial de tumor con hallazgos posoperatorios de adenoma con componentes somatotropo escasamente granulado, lactotropo densamente granulado y células sugerentes de adenoma tirotrófico. La conclusión fue de adenoma plurihormonal (Figura 2), la inmunohistoquímica mostró prolactina (+), ACTH (-), LH (-), GH (+), TSH (+), P53 (-), Ki67 3%, alteración y ruptura de red de reticulina y PAS negativo.
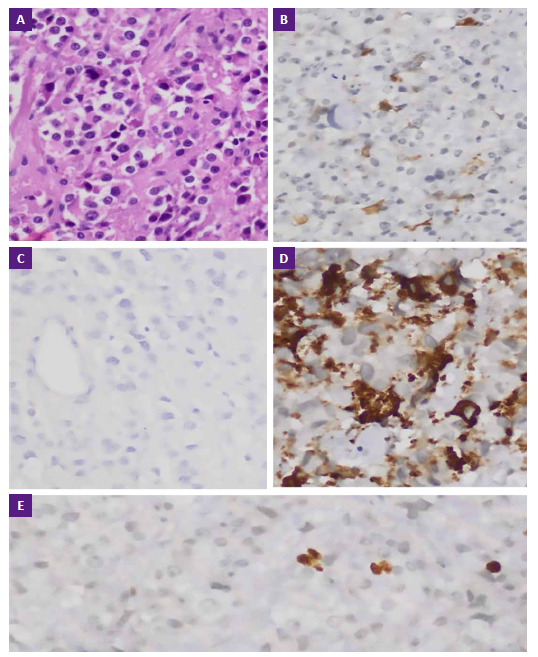
Figura 2. Estudio histopatológico de pieza operatoria: A) células hipofisiarias neoplásicas con núcleos acidófilos redondeados con variabilidad en el tamaño nuclear, algunos con nucleolo prominente y citoplasma eosinófilo. Cromatina en sal y pimienta; tinción de hematoxilina - eosina x400. B) Inmunohistoquímica (IHQ) positivo para hormona de crecimiento. C) IHQ positivo para hormona estimulante de la tiroides (TSH). D) IHQ positivo para hormona estimulante de la tiroides. E) El índice de proliferación Ki67 fue de 3% en las células tumorales; IHQ x400.
En el posoperatorio cursó con edema cerebral, insuficiencia respiratoria, infección de herida operatoria e hipertiroidismo secundario descompensado, por lo que fue reintervenido 12 días mediante una craneotomía descompresiva. El paciente presentó tormenta tiroidea sin respuesta a tratamiento. Se decidió la realización de plasmaféresis para tratamiento de la tirotoxicosis. Debido a dificultad del tratamiento de la tirotoxicosis, se decidió tiroidectomía total más traqueostomía, posterior a ello, cursó con evolución favorable con secuela de hemiparesia derecha.
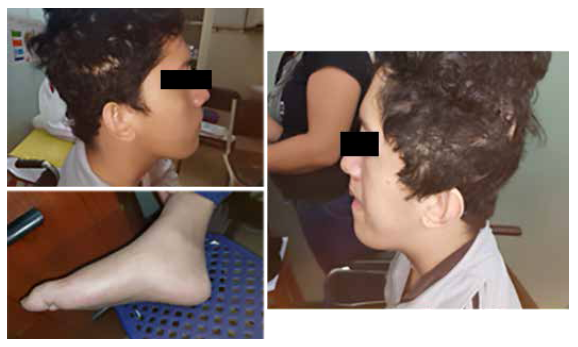
El paciente fue dado de alta hospitalaria y referido al Instituto Nacional de Enfermedades Neoplásicas, donde recibió radioterapia por 18 sesiones. Tras 2 años de la cirugía y un año de la radioterapia el paciente se encuentra en seguimiento por consultorio externo donde recibe terapia de reemplazo hormonal por hipopituitarismo con prednisona, levotiroxina y enantato de testosterona, además de usar de cabergolina y en espera de acceder a uso de análogos de somatostatina (Figura 3).
DISCUSIÓN
Los tumores pituitarios se encuentran hasta en 10% de la población, pero solo 2% de estos tumores son clínicamente relevantes, ya sea por su producción hormonal o por el efecto de masa sobre las estructuras adyacentes 4. Los tumores pituitarios gigantes son definidos por un diámetro tumoral mayor a 4 cm, siendo en su gran mayoría (68,8%) no funcionantes y más comúnmente reportados en hombres 5. Dentro de los tumores gigantes funcionantes, el prolactinoma es el más común (54,2%), seguido por somatotropinoma (29,2%), gonadotropinoma (8,3%), corticotropinoma (4,2%) y tirotropinoma (4,2%).
Nuestro paciente presentó un adenoma plurihomonal con exceso de hormona de crecimiento, prolactina y TSH. Los tumores pituitarios plurihormonales representan 10 a 15% de los tumores pituitarios. Las combinaciones hormonales más comunes incluyen hormona de crecimiento (GH), prolactina (PRL) y/o una hormona glicoproteica (TSH, LH, FSH) 6,7. En un estudio que incluyó a 1628 adenomas pituitarios operados, dentro de los plurihormonales 85% secretaban GH+TSH 8. En otra revisión de Beck y col, las asociaciones más frecuentes de los tumores productores de TSH (tirotropinomas) eran generalmente con hormona de crecimiento (16%) y prolactina (10,4%) 9.
La presencia de un tirotropinoma gigante es de destacar en nuestro paciente. Los tirotropinomas tienen mecanismos moleculares de formación poco conocidos. Dentro de los protooncogenes se han encontrado sobreexpresión del factor de trascripción Pit-1, involucrado también en la diferenciación celular y expresión de los genes de PRL, GH y TSH 9. Existen reportes que notifican que la resolución quirúrgica es menor en adenomas plurihormales por alteración de Pit-110,11.
La asociación de un tirotropinoma y un exceso de hormona de crecimiento se ha reportado previamente. En un estudio retrospectivo se evaluó 21 adenomas pituitarios productores de TSH y GH, en estos casos la distribución de edad fue bimodal con picos a los 20 y 40 años, y una razón varones/ mujeres de 3:2. Todos los tumores fueron macroadenomas, 23% fueron gigantes, la destrucción del piso de la silla turca ocurrió en un tercio de los pacientes y la invasión del seno cavernoso en 57,1%. El bocio difuso estuvo presente en 42,9% y los nódulos tiroideos en 47% 12. Nuestro paciente tenía una presentación clínica similar.
Como se ha mencionado nuestro paciente presentó un adenoma plurihormonal con componentes de tirotropinoma, exceso de hormona de crecimiento y prolactina, pero además tuvo biomarcadores de agresividad como el ki67. En los adenomas hipofisiarios la positividad de ki67 suele ser baja (< 3%) y una elevación ≥ a 3% se correlaciona con mayor velocidad de crecimiento, invasión y recurrencia tumoral como se presentó en nuestro caso (3%). A diferencia de otros tumores neuroendocrinos, los de pituitaria no se estratifican en grados según su índice de proliferación Ki67; esto dado que existen biomarcadores mas precisos de comportamiento agresivo como la estirpe celular, factores de transcripción, entre otros.
Dentro de las características de agresividad que se han encontrado a nivel histológico están la presencia de un adenoma lactotrófico densamente granulado y somatotrófico escasamente granulado lo que puede explicar el comportamiento agresivo de este tumor neuroendocrino a nivel hipofisiario e incluso que no pueda responder plenamente al tratamiento con agonistas dopaminérgicos o análogos de somatostatina 13. Lamentablemente en nuestro medio no se cuentan con estudios de inmunohistoquímica de factores de transcripción para definir el linaje celular. El conocimiento de biomarcadores de agresividad en los tumores neuroendocrinos de la pituitaria nos muestran la heterogeneidad de la naturaleza de estos y la necesidad de un enfoque individualizado 14.
El abordaje en nuestro paciente incluyó resección quirúrgica como primera línea de tratamiento del adenoma hipofisiario, manejo concomitante del hipertiroidismo secundario mediante tiroidectomía y tratamiento coadyuvante con radioterapia en el posoperatorio, así como el uso de agonistas dopaminérgicos. Otros tipos de terapias coadyuvantes como análogos de somatostatina no se encontraban disponibles.
En relación con el tratamiento y resultados de tumores productores de TSH, una revisión sistemática encontró que la remoción total no es posible siempre si hay invasión al seno cavernoso y si era un macroadenoma. La terapia adyuvante incluyó radioterapia encontrándose que en 15 estudios se logró remisión bioquímica en el 65,9% de pacientes 15. Los hallazgos y manejo indicados fueron semejantes a los de nuestro caso.
En un estudio en 21 pacientes con tumores pituitarios secretores de TSH y GH, la cirugía fue la primera opción en 19, 14 recibieron ligandos de somatostatina previos a la cirugía y 4 pacientes recibieron tratamiento adicional que incluía radioterapia. En el seguimiento antes de los dos años, el 23,8% de pacientes lograron remisión a largo plazo en el eje GH/IGF-1 y el 38% lograron remisión en el corto plazo, la no remisión o recaída se encontró en 38% 12.
Otro fármaco, no aplicado en este caso, para el tratamiento adyuvante de tumores pituitarios agresivos y carcinomas de primera línea dentro de la quimioterapia es la temozolomida (TMZ) pudiendo ser usada previa a la radioterapia o incluso previo a la re-operación 14. La combinación con radioterapia puede tener un efecto sinérgico en aquellos tumores con Ki67 superior al 10%, con una buena respuesta en el 75% comparado con el 40% de respuesta en monoterapia con TMZ 2. Otras terapias coadyuvantes como los inhibidores de puntos de control inmunitario como anticuerpos antiCTLA-4 y anticuerpos anti-PD-1/PD-L1 se han evaluado en pacientes con tumores pituitarios agresivos y carcinomas pituitarios encontrando mejor repuesta en carcinomas pituitarios 2.
Es importante destacar que el paciente presentó tormenta tiroidea como complicación de la cirugia descompresiva. Aunque la tormenta tiroidea suele desarrollarse en pacientes con hipertiroidismo no tratados de larga duración, ésta puede precipitarse por un evento agudo como una cirugía tiroidea o no tiroidea, traumatismo, infección, una carga aguda de yodo o parto. En los pacientes hospitalizados la infección es el desencadenante más común y hasta en el 25 a 43% de los casos no hay un factor precipitante identificable 17.
En relación con el tratamiento del hipertiroidismo secundario en nuestro paciente se realizó la tiroidectomía. Hasta hace algunos años, se creía que similar al síndrome de Nelson que se producía tras la adrenalectomía, podía ocurrir lo mismo tras la tiroidectomía, sin embargo, aun cuando la experiencia es escasa en los pocos casos reportados no se encontró crecimiento de los tirotropinomas 18. En el caso presentado, debido a la inestabilidad clínica del paciente se optó por esta alternativa con buena respuesta.
En conclusión, en el presente caso se evaluó a un paciente con un tumor neuroendocrino plurihormonal con clínica de acrogigantismo asociado a tirotoxicosis. Los tumores neuroendocrinos de la pituitaria con biomarcadores de agresividad pueden tener un comportamiento agresivo que los pueda hacer refractarios al tratamiento convencional de tipo farmacológico, quirúrgico o de radioterapia. A futuro la detección de biomarcadores relacionados al origen del exceso de producción uni o plurihormonal puede llevar a un diagnóstico y tratamiento oportuno e individualizado además de una apropiada consejería genética.

