











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO  uBio
uBio 
 Permalink
PermalinkINTRODUCCIÓN
La crianza de alpacas es una de las actividades de mayor importancia e impacto en el desarrollo socioeconómico de la población altoandina del Perú, por su capacidad de adaptación a condiciones ambientales extremas, por ser una fuente alimenticia proteica para la población humana, y por su producción de fibra.
Los polimorfismos de nucleótido simple (PNSs) son variaciones en la secuencia de ADN que afectan una base nitrogenada. Están siendo muy utilizados como marcadores moleculares con la finalidad de investigar procesos genéticos evolutivos, estudios de asociación a caracteres de interés productivo y para incrementar el conocimiento en especies no modelo donde no se conocen detalles sobre su genoma (Seeb et al., 2011).
Existen estrategias para estimar la localización y ordenamiento de marcadores moleculares a lo largo del genoma de cualquier especie, siendo una de ellas el uso de paneles de células híbridas irradiadas, cuyo fundamento es la fusión de células de la especie de interés (donadora), conteniendo fragmentos de ADN causados por la irradiación, con células inmortales de otra especie (receptora) que retiene los fragmentos de ADN donados. El análisis de retención de marcadores moleculares en estas células híbridas irradiadas determina el ordenamiento de marcadores moleculares en grupos ligados para la especie de interés (Faraut et al., 2009). El análisis de co-segregación de PNSs en las células híbridas irradiadas se facilita mediante el genotipaje de cada uno de los clones irradiados con una micromatriz de PNSs, que permite hacer genotipados de miles de PNSs en un solo análisis (Wiggans et al., 2017).
Johnson y Perelman (2007) reportaron la creación de un panel celular de radiación híbrida (5000 Rad) de una alpaca macho llamada "Limerick", conformado por 92 clones. El panel está compuesto por clones con frecuencias de retención mayores al 40%, donde se llegaron a mapear 428 marcadores en base a la homología encontrada con ZooFISH existente entre los cromosomas de dromedario, bovino y humano (Balmus et al., 2007). De la misma manera, Perelman et al. (2018) reportaron la creación de dos paneles híbridos irradiados (5000 y 15000 Rad) para el dromedario. El primero está constituido por 93 clones y su frecuencia de retención es de 47.7%, y el segundo compuesto por 90 clones y una frecuencia de retención de 39.9%. Estos paneles de radiación híbrida constituyen un recurso importante para la construcción de mapas genómicos de mediana y alta resolución en camélidos, respectivamente.
Se cuenta con un mapa preliminar citogenético integrado del genoma de alpaca, el cual está conformado por 230 marcadores moleculares ordenados a lo largo de los 37 pares de cromosomas de la alpaca (Ávila et al., 2014). Asimismo, 86 genes de alpacas han sido mapeados en el dromedario, observándose localizaciones sinténicas entre ambos, confirmando que ambas especies son genómicamente muy similares. Mendoza et al. (2019) localizaron seis genes candidatos para el crecimiento de fibra (COL1A1, CTNNB1, DAB2IP, KRT15, KRTAP13-1, y TNFSF12) y cinco genes para el color de fibra (ALX3, NCOA6, SOX9, ZIC1, y ZIC5). También se identificaron 48 PNSs ubicados en regiones intrónicas y exónicas de 22 genes de queratina (Fernández et al., 2019).
No se han diseñado micromatrices para análisis genómico en alpacas, por lo que existe la posibilidad de utilizar micromatrices ya existentes en otras especies para identificar PNSs en alpacas. Estos PNSs serían conservados entre alpaca y bovino. Estudios en venados (Haynes y Latch, 2012) y bisonte (Pertoldi et al., 2010) han logrado identificar PNSs con el uso de la micromatriz de bovino, evaluando más de 50 000 PNSs (BovineSNP50 BeadChip, Illumina). Sin embargo, cuando se analizan dos especies hay que considerar las distancias evolutivas entre ellas, dado que podría existir una relación inversa entre distancia evolutiva y el número de PNSs conservados. Por lo tanto, el uso de micromatrices de bovino para la identificación de PNSs comunes con la alpaca podría ser beneficioso para incrementar el número de PNSs de alpaca.
Por este motivo, el objetivo del estudio fue identificar PNSs conservados entre bovino y alpaca y desarrollar un mapa físico preliminar de estos PNSs usando un panel celular híbrido irradiado alpaca/hámster y una micromatriz de genotipado de alta densidad de bovino.
MATERIALES Y MÉTODOS
Se utilizó como material experimental un panel de 92 clones celulares híbridos irradiados alpaca/hámster y cuatro muestras control adicionales al panel que contienen ADN genómico de alpaca Huacaya macho (Limmerick), alpaca huacaya hembra (Carlotta), la línea celular de hámster A23 y una mezcla de alpaca macho y hámster en una proporción de 1:10, respectivamente. Estas 96 muestras fueron genotipadas con la micromatriz de alta densidad para bovinos (BovineHD BeadChip-Illumina) que contiene 777 962 PNSs.
Los resultados del genotipado de las cuatro muestras controles fueron analizados con los programas GenomeStudio, Excel y R. El parámetro utilizado para seleccionar PNSs fue la frecuencia de señal positiva (Call Frequency) equivalente a 1. Solo se aceptaron PNSs que fueron identificados en las dos muestras de alpaca con frecuencia 1, eliminando del análisis el resto de PNSs que no cumplieron con este criterio. Las dos muestras de ADN de alpaca, la muestra de hámster y la muestra mezcla de alpaca macho/ hámster 1:10 se evaluaron de forma independiente y se generaron bases de datos en formato .txt y .xlsx. La información obtenida de estas muestras sirvió para, por comparación, retener PNSs con frecuencia 1 en las muestras de ADN de alpaca y hámster, para luego eliminar los conservados comunes entre ambas muestras y solo se tabularon los PNSs únicos en alpaca.
Los resultados del genotipado de los 92 clones celulares híbridos irradiados alpaca/ hámster fueron evaluados en el GenomeStudio 2011.1 (Genotyping Module v. 1.9.4, Illumina, USA), considerando la frecuencia de señal positiva >0.20 y <0.80. Luego los PNSs comunes con los que estuvieron presentes en el ADN de alpaca y ausentes en el ADN de hámster evaluados anteriormente, fueron tabulados con su genotipo respectivo. Finalmente, como el panel fue realizado con el ADN de una alpaca, el genotipado de cada PNS en los 92 clones celulares híbridos podría ser solo homocigoto (AA o BB), o heterocigoto (AB) o ausencia de señal (no genotipado, NC); donde ningún PNS debe haber sido homocigoto para ambos alelos o la combinación de homocigotos con heterocigotos. Por esta razón, solo se consideraron los PNSs cuyos genotipos fueron similares en todas las muestras evaluadas. De esta manera se eliminaron PNSs en los que se asume señal positiva errónea (falsos positivos).
Para calcular las distancias y el orden de los PNSs encontrados previamente se utilizó el programa Carthagene (Schiex et al., 2009) para lo cual se tuvo que transformar la información obtenida al formato MapMaker, codificando con el valor 1 los genotipos AA, AB y BB, los no genotipados (NC) con el valor 0, en un archivo de texto delimitado por tabulaciones. Luego en base a la localización cromosómica de PNSs bovinos, los PNS positivos de alpaca se catalogaron en 30 grupos, facilitando de esta manera el análisis de ligamiento con el programa Carthagene. Una vez identificados los grupos de PNSs ligados se crearon tablas de datos donde se registraron con su secuencia bovina total de 121 nucleótidos, ubicándose el polimorfismo en el medio. Esta información fue extraída de la base de datos de PNSs del BovineHD BeadChip usando el programa GenomeStudio.
De la misma manera, se crearon diferentes tablas que incluían la secuencia de 20 y 25 nucleótidos anteriores al nucleótido polimórfico en dirección 5' 3', ubicándose de esta manera el polimorfismo como el nucleótido final de la secuencia. El genoma de referencia Vicugna_pacos-2.0.2 del NCBI (National Center for Biotechnology Information) fue descargado en formato FASTA del GenBank (GCF_000164845.1) y procesado con el software REPEATMASKER, con la finalidad de eliminar secuencias repetitivas del genoma de la alpaca. Este último file se subió a la plataforma Galaxy (Cock et al., 2015) alojado en el Minnesota Supercomputing Institute (University of Minnesota) con la finalidad de localizar los PNSs bovinos que resultaron positivos en alpaca en los scaffolds correspondientes al genoma de alpaca, mediante el programa BLAST y los comandos SHORT BLAST a un valor esperado (E-value) de 0.05 (NCBI, 2016).
El software Carthagene utiliza un modelo probabilístico denominado Modelo Oculto de Markov y para el cálculo de la frecuencia de retención de los PNSs evaluados utiliza un estimador de máxima verosimilitud cuyo algoritmo es el de esperanza-maximización (EM). Esta probabilidad calculada es transformada a un parámetro llamado LOD score (logaritmo de las probabibidades), el cual indica cuando dos loci o marcadores están ligados y se van a heredar juntos. Un LOD score de 3 indica que por cada 1000 análisis uno (1) sea diferente de lo observado al azar (De Givry et al., 2004).
Para el caso de la localización de los PNSs en el genoma de la alpaca se calculó el E-value, el cual hace referencia al número de veces (probabilidad) que ocurran excelentes alineamientos comparados a que ocurran al azar en una base de datos. Las alineaciones biológicamente significativas, que indican que las secuencias de ADN son significativamente similares, tienen E-value<1 (Pesvener, 2015).
RESULTADOS
El esquema de análisis e identificación de PNSs que emitieron señal positiva y estuvieron presentes solo en el genoma de alpaca se realizó de la manera que fue descrito por Mamani et al. (2017). Brevemente, de los 777 962 PNSs analizados mediante la micromatriz de alta densidad, 294 165 PNSs fueron positivos en las dos muestras control de alpaca, lo que representa el 37.81% del total. En la muestra de hámster se identificaron 453 699 PNSs, y luego de realizar filtrados y eliminar los PNSs comunes con alpaca, se encontró un total de 72 964 PNSs conservados entre la alpaca y el bovino. De la misma manera, la muestra que contenía ADN de alpaca y hámster en la proporción 1:10 se comparó con el resto de las muestras control para poder descartar PNSs falsos positivos y de esta manera seleccionar con mayor astringencia PNSs específicos de alpaca. Esta última comparación identificó 50 686 PNSs, que representan el 6.5% del total.
De los 50 686 PNSs identificados, solo quedaron 2924 PNSs que cumplieron el requisito de ser AA y NC o BB y NC o AB y NC. La distribución de estos PNSs por cromosoma bovino se muestra en la Figura 1. Cada uno de estos grupos de PNSs fue evaluado con el programa Carthagene y los resultados se muestran en el Cuadro 1, donde se identifican 33 grupos ligados con un total de 216 PNSs presentes en el genoma de la alpaca. Los mapas de referencia (framework maps) generados con el orden, nombre y distancias entre PNSs se presentan en las Figura 2, Figura 3 y Figura 4.
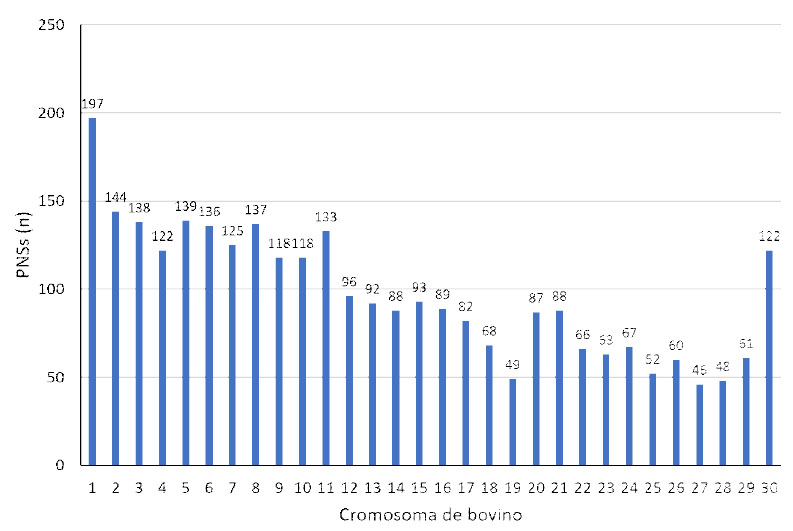
Figura 1 Distribución de polimorfismos de nucleótido simple (PNSs) conservados entre alpaca y bovino
Cuadro 1 Identificación de grupos de ligamiento en base a su orden, distancia, logaritmo de probabilidades y probable homología entre cromosomas de bovino y alpaca
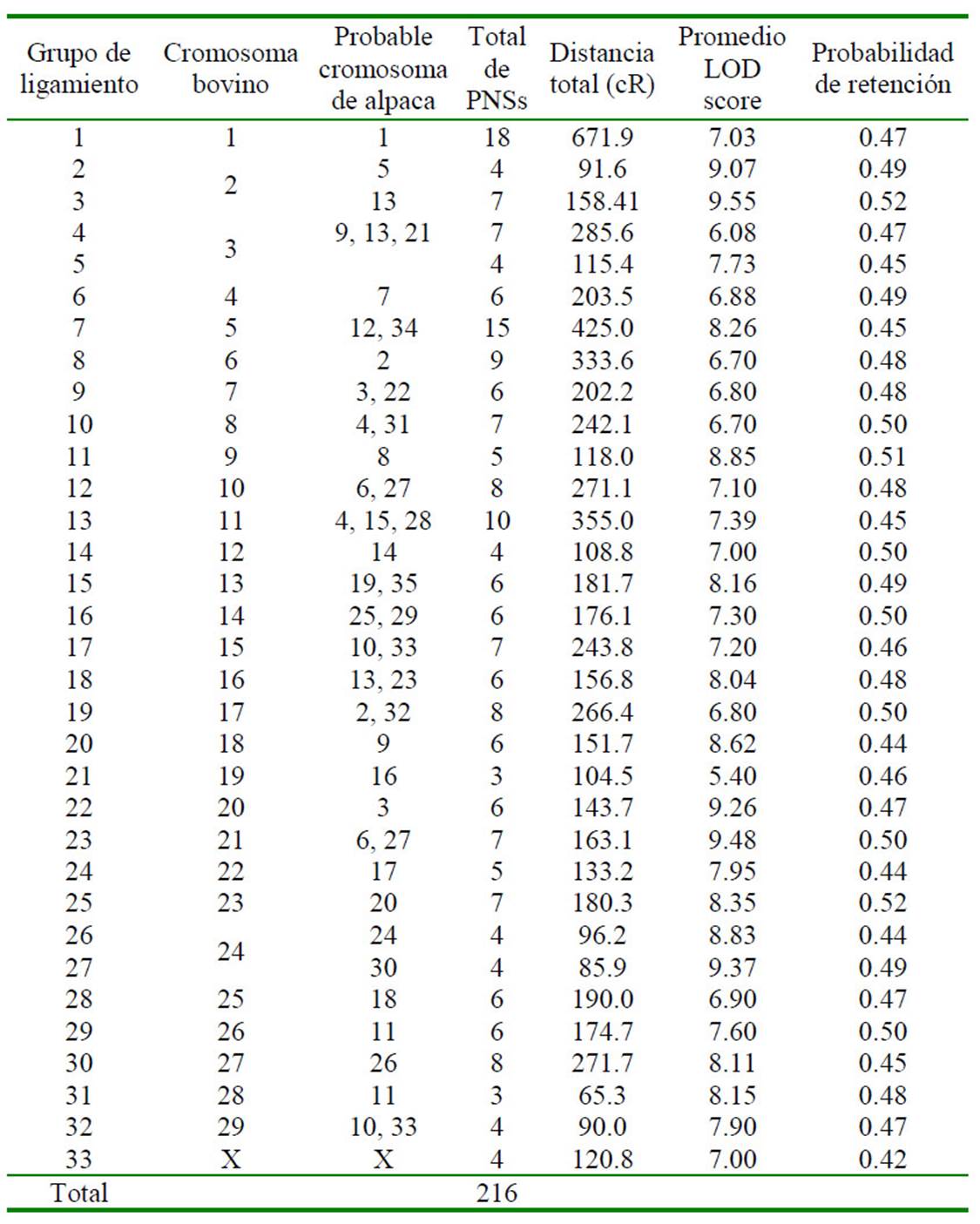

Figura 2 Mapas de referencias con los grupos de ligamiento (GL) del 1 al 11, con el orden y distancias relativas

Figura 3 Mapas de referencias con los grupos de ligamiento (GL) del 12 al 22, con el orden y distancias relativas

Figura 4 Mapas de referencias con los grupos de ligamiento (GL) del 23 al 33, con el orden y distancias relativas
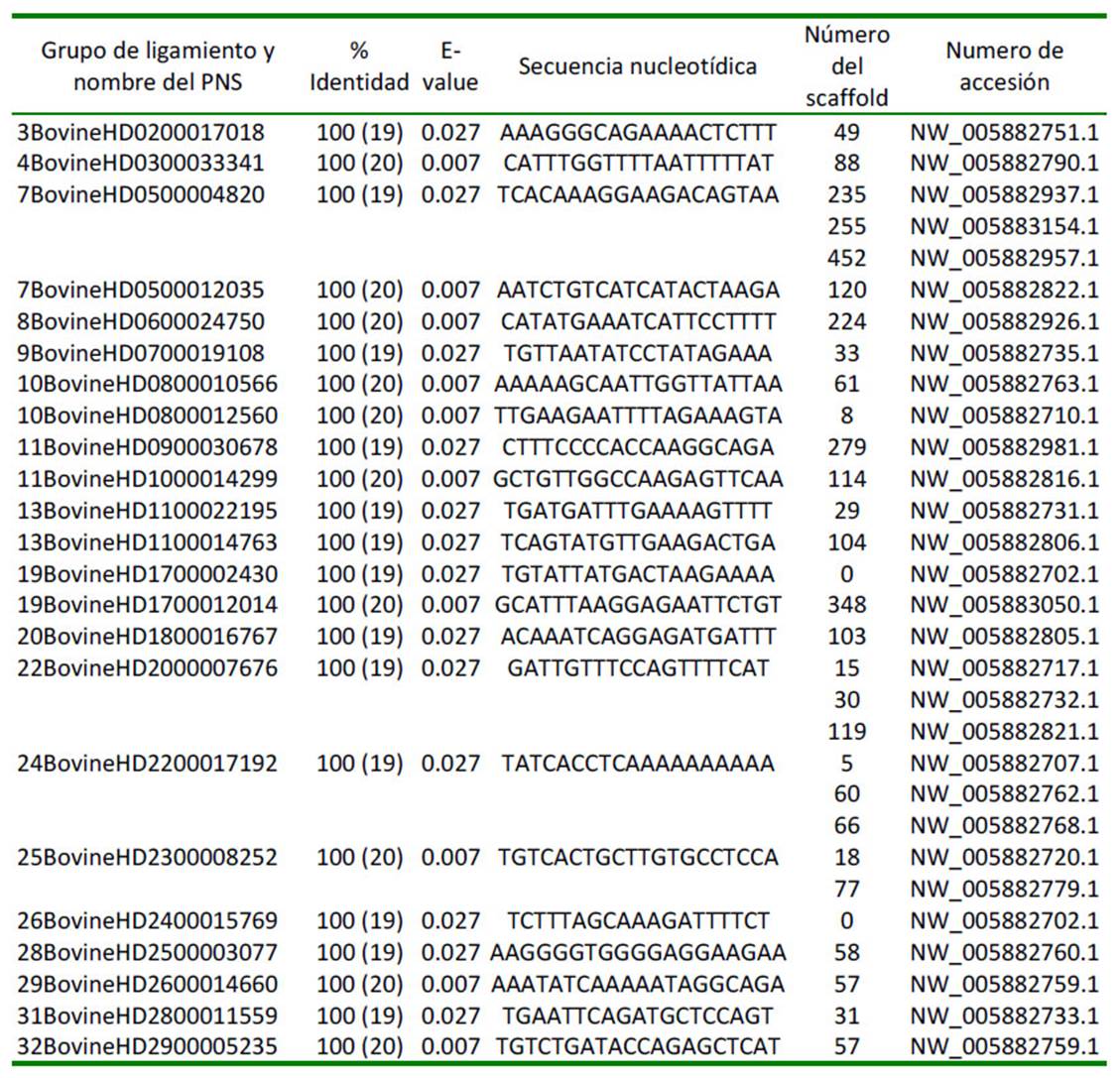
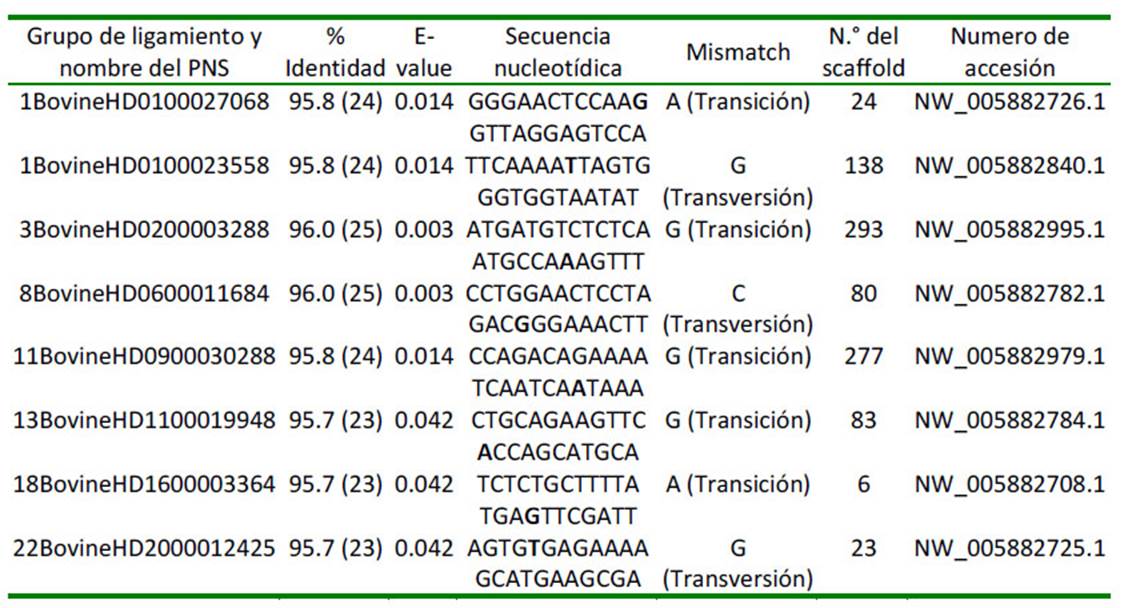
El Cuadro 2 y Cuadro 3 muestran la ubicación de 23 y 8 PNS por comparación de las secuencias de 20 y 25 nucleótidos, respectivamente, en el Vicugna_pacos-2.0.2, todos con E-value <0.05. Se consideró este valor porque es más restrictivo al momento de inferir si los alineamientos son producto del azar (Pesvener, 2015).
Cuadro 2 Secuencias de polimorfismos de nucleótido simple (PNSs) localizadas en el Vicugna_pacos-2.0.2 con un tamaño de 20 nucleótidos (E-value <0.05)
DISCUSIÓN
Con el fin de incrementar el número de marcadores moleculares identificados para especies con información genómica insuficiente, se han usado micromatrices de marcadores moleculares disponibles de otras especies que son evolutivamente cercanas. Por ejemplo, en venados usando la micromatriz de 50K disponible para bovinos se pudo obtener 38.7% de PNSs conservados entre estas especies (Haynes y Latch, 2012). Este resultado concuerda con el valor de 37.81% de PNSs que emitieron señal positiva, utilizando la micromatriz de 777K, reportada en el presente estudio. Sin embargo, en otro estudio realizado en dos poblaciones de bisonte se reportó 96.7 y 98.7% de PNSs conservados usando la micromatriz bovina de 50K (Pertoldi et al., 2010). Las distancias evolutivas entre el venado-bovino (Hassanin y Douzery, 2003), camélidos-bovinos (Wu et al., 2014) y bisonte-bovino (MacEachern et al., 2009) fueron estimadas en 25.1 a 30.1, 42.7 y 1.2 a 2.1 millones de años, respectivamente. Por lo tanto, al existir una mayor divergencia evolutiva entre camélidos y bovinos, se espera obtener una menor cantidad de PNSs identificados usando la micromatriz del bovino.
Bertolini et al. (2016) detectaron 10.5% PNSs en ocho alpacas usando la misma micromatriz de 777K empleada en la presente investigación, donde se detectó el 6.5% del total de PNSs analizados. Las razones para estas diferencias podrían ser porque en este estudio se utilizó el genoma de una alpaca en la construcción de los clones de células híbridas irradiadas y se excluyeron los comunes con hámster.
De los 50 686 PNSs detectados en el ADN de alpaca y ausentes en el hámster se utilizaron solo 2924 PNSs para el análisis de ligamiento. Esta disminución se debió al criterio que se impuso para incrementar la astringencia del análisis descartando los posibles falsos positivos. En el Cuadro 1 se presenta la relación de los 33 grupos de ligamiento que incluyen 216 PNSs de los 2924 resultantes del análisis con el programa Carthagene. Esta disminución podría explicarse por el nivel de radiación (5000 rad) utilizado para generar el panel celular híbrido irradiado, porque a menor dosis de radiación los fragmentos de ADN que se generan son más grandes y por lo tanto el análisis de ligamiento de un número muy grande de marcadores PNSs tiene una menor resolución. La decisión de analizar grupos de PNSs por cromosoma de bovino también disminuyó el número total de PNSs que podrían ser parte de los grupos ligados.
Según los estudios realizados por Balmus et al. (2007), existen correspondencias cromosómicas entre el bovino y dromedario que pueden ayudar a ubicar cromosómicamente marcadores moleculares, tal como Ávila et al. (2014) que evidenciaron con sus estudios citogenéticos y mapeo por hibridación fluorescente in situ (FISH) que existe un escaso o ningún desarreglo cromosómico entre las especies de camélidos.
En el Cuadro 1 se muestra la probable homología entre cromosomas del bovino y la alpaca, donde se aprecia que el cromosoma 2 del bovino tiene sintenia compartida con los cromosomas 5 y 13 de la alpaca, el cromosoma 3 del bovino tiene sintenia compartida con los cromosomas 9, 13 y 21 de la alpaca y el cromosoma 24 del bovino tiene sintenia compartida con los cromosomas 24 y 30 de la alpaca. En tanto, estos cromosomas de alpaca han sido clasificados como subtelocéntricos o acrocéntricos (5, 9, 13), submetacéntricos (21, 24) y metacéntrico (30), por lo que explicaría la presencia de dos grupos de ligamiento en alpacas que podrían estar presentes en los brazos cromosómicos p y q respectivamente (Ramos, 2014).
Cabe resaltar que este trabajo sería el primero en identificar grupos de ligamiento usando la micromatriz de alta densidad del bovino para el genoma de alpaca. La metodología de mapeo por radiación híbrida ha permitido incrementar el número de marcadores moleculares en especies como el vacuno (Zimin et al., 2009), cerdo (Servin et al., 2012) y caprino (Du et al. 2014), debido a que en combinación con la información de secuenciamiento y otras metodologías de mapeo físico producen mayor precisión en el ensamblaje de genomas de referencia para lograr mapas de mayor resolución a nivel cromosómico. Por esta razón y considerando que en alpacas aún no existe una micromatriz de ADN para identificar PNSs, su utilidad será muy importante para complementar la información de su genoma.
Las distancias medidas en cR en los 33 grupos de ligamiento varían desde los 65.3 cR hasta los 671.9 cR. Para efectos del presente estudio son mediciones relativas entre PNSs en los grupos de ligamiento respectivos. Por lo tanto, la resolución expresada como kilobases (Kb) por cR no fue estimada en el presente trabajo. Sin embargo, en otras especies, como cerdos, se calculó una resolución de 8.6 Kb/cR y 5.3 Kb/cR usando paneles de 7,000 y 12,000 rads, respectivamente (Servin et al., 2012). En el caso del caprino se estimó una resolución del mapa de 32.6 Kb/cR usando el panel de 5000 rad (Du et al., 2014). Como se aprecia en los casos del porcino y caprino, a medida que se incrementa el nivel de radiación en la elaboración del panel, la resolución del mapa generado va a ser mayor, entonces la cantidad de marcadores moleculares que se podrían mapear con mejor precisión seria también mayor.
Al momento de analizar grupos de marcadores y verificar si están o no ligados se estima la estadística LOD >3.0 y los valores resultantes en los 33 grupos de ligamiento estuvieron por encima del LOD >6, lo que indica que existen altos niveles de probabilidad de que el orden y las distancias sean correctos. En el presente estudio se decidió usar el valor LOD=6 para incrementar la astringencia de asociación entre los marcadores que componen el mapa, lo que a su vez redujo la posibilidad de incrementar el número de marcadores por grupo de ligamiento.
Los porcentajes de identidad mostrados en el Cuadro 2, producto de secuencias query de 20 nucleótidos, son calculados sumando las correspondencias idénticas entre secuencias alineadas y dividiendo entre el número total de bases alineadas. En el presente caso, todos los PNSs en los mapas desarrollados fueron del 100%; sin embargo, los emparejamientos idénticos (matches) en algunos casos fueron de 19 nucleótidos y otros de 20 (Klug et al., 2016). Los valores del Evalue en los 23 PNSs fueron menores a 0.05; por lo tanto, los alineamientos son significativos y tienen una mayor probabilidad que las secuencias ubicadas en el Vicugna_pacos-2.0.2 no sean productos del azar.
El Cuadro 3 muestra un total de ocho PNSs identificados en el Vicugna_pacos-2.0.2 al analizar las secuencias query de 25 nucleótidos. A diferencia del Cuadro 2, los porcentajes de identidad son mayores al 95%, en donde aparecen alineamientos de 23 hasta 25 nucleótidos y la presencia de un mismatch (no emparejamiento) en todos los casos. En la columna de mismatch se considera el nucleótido que no alineó correctamente y se verifica si el cambio de base fue por transición o transversión. Los valores del Evalue en los ocho PNSs fueron menores a 0.05, por lo tanto, los alineamientos son significativos.
Los resultados del presente análisis solo ubicaron 23 y 8 PNSs de los 216 PNSs en el Vicugna_pacos-2.0., que se puede deber a una serie de factores como: la mayor divergencia evolutiva que existen entre el vacuno y la alpaca (Wu et al., 2014) y que también explicaría la aparición de mismatch entre ambas especies, el valor más restringido del E-value que se consideró, la poca información que se tenía en cuanto al número exacto de nucleótidos que reacciona con la micromatriz y hace positiva su aparición en los resultados, y a la cobertura de 22X que no cubre el genoma completo de la alpaca.
CONCLUSIONES
Se identificaron 50 686 polimorfismos de nucleótido simple utilizando la micromatriz de alta densidad del bovino y con un nivel más alto de astringencia, solo 2924 se estimaron como verdaderos PNSs de alpacas. Con este último dato se desarrolló un primer mapa referencial que resultó en 33 grupos de ligamiento que incluyen 216 PNSs, de los cuales fueron ubicados 31 PNSs en el genoma referencial evaluado

