











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
Cada vez más países de América Latina aplican sus políticas de medicamentos con el objetivo de garantizar la disponibilidad y accesibilidad a medicamentos seguros, eficaces y de calidad, a fin de satisfacer las necesidades de salud de la población 1, y promover la sustitución de medicamentos basándose en la promoción de genéricos a un precio más bajo 2.
Un medicamento genérico es el medicamento que ha demostrado ser equivalente al innovador y es intercambiable con él en la práctica clínica. Por esta razón, el concepto de intercambiabilidad cobra importancia y se ha convertido en un requisito para autorizar la comercialización de los genéricos 3.
El reglamento que regula la intercambiabilidad de medicamentos en el Perú (Decreto Supremo 024-2018-SA) permitirá garantizar la disponibilidad de medicamentos genéricos seguros, eficaces y de calidad 4.
Este artículo aborda aspectos relacionados con los genéricos en el contexto peruano, asimismo describe la implementación de la intercambiabilidad de medicamentos, centrándose en los requisitos regulatorios y las perspectivas de la aplicación de la normativa vigente.
ANTECEDENTES Y CONTEXTO PERUANO
El término «medicamento genérico» tiene diferentes significados según la jurisdicción en la que se utilice 5. La Administración de Medicamentos y Alimentos (FDA, por sus siglas en inglés) de los Estados Unidos, define que un genérico es idéntico a un medicamento de marca en forma de dosificación, seguridad, concentración, vía de administración, calidad, características de rendimiento y uso previsto 6. La Agencia Europea de Medicamentos (EMA, por sus siglas en inglés), lo define como el producto que presenta la misma composición cualitativa y cuantitativa en principios activos, la misma forma farmacéutica que el medicamento de referencia, y cuya bioequivalencia ha sido demostrada mediante estudios de biodisponibilidad adecuados 7.
En América Latina, por la diversidad de definiciones, la Organización Mundial de la Salud (OMS), en lugar de «medicamento genérico» utiliza el término «producto farmacéutico multifuente», el cual no se encuentra asociado con el vencimiento de las patentes ni con el cumplimiento de la equivalencia terapéutica; por el contrario, es definido como equivalente o alternativa farmacéutica que puede ser o no equivalente terapéutico 3. A este respecto, en el Perú no existe la figura del medicamento genérico como tal; por el contrario, se ha adoptado otra definición, es decir, como el producto farmacéutico cuyo nombre coincide con la Denominación Común Internacional del ingrediente activo y no se identifica con el nombre de marca 8 , 9.
La intercambiabilidad de medicamentos en el Perú comenzó con la Política Nacional de Medicamentos del 2004, en la que se apoya la aplicación gradual de estudios de biodisponibilidad y bioequivalencia de medicamentos de alto riesgo sanitario 10. La Ley 29459 del 2009, establece que los medicamentos estén sujetos a estudios de intercambiabilidad 9. En el 2011 se aprobó el reglamento de la citada ley, Reglamento para el registro, control y vigilancia sanitaria de productos farmacéuticos, dispositivos médicos y productos sanitarios (D. S. 016-2011-SA), en el que se incluye el requisito de presentar estudios de equivalencia terapéutica en el procedimiento de inscripción y reinscripción para el registro sanitario de medicamentos de categoría 1 y 2 11. En el 2015 se publicó el proyecto del reglamento que regula la intercambiabilidad de medicamentos a efectos de recibir sugerencias y comentarios 12, el cual se aprobó el 15 de septiembre del 2018, mediante D. S. 024-2018-SA, y entró en vigencia seis meses después 4.
IMPLEMENTACIÓN DE LA INTERCAMBIABILIDAD DE MEDICAMENTOS
Con la entrada en vigencia del reglamento de intercambiabilidad, la demostración de la equivalencia terapéutica de los medicamentos genéricos constituye una «nueva exigencia» para obtener el registro sanitario en el Perú. En su elaboración se tuvieron en cuenta las directrices de la OMS, la EMA, Health Canada y la FDA, y consta de 5 títulos, 27 artículos y 9 disposiciones complementarias finales, en ella se define el procedimiento para establecer la intercambiabilidad siguiendo los criterios de riesgo sanitario de los medicamentos y la gradualidad en la presentación de los estudios 4.
La equivalencia terapéutica se determina mediante estudios in vivo (estudios farmacocinéticos comparativos o bioequivalencia, estudios farmacodinámicos y ensayos clínicos comparativos) o estudios in vitro (bioexención) entre el producto de prueba y el de referencia. El estudio de bioequivalencia es el más utilizado 13 , 14.
El medicamento de referencia o comparador es, en general, el innovador que fue autorizado por primera vez para su comercialización (generalmente como medicamento patentado); para el que se ha establecido la seguridad, eficacia y calidad; y con el que se pretende que el producto de prueba sea intercambiable en la práctica clínica 15. La autoridad reguladora es la encargada de seleccionar el medicamento de referencia 16. Para ello, la Dirección General de Medicamentos, Insumos y Drogas (DIGEMID) ha establecido el orden de prioridad a seguir, comenzando por: 1) el innovador fabricado en el primer país de origen; 2) el innovador fabricado en un país de origen alterno; 3) el producto de referencia de las listas de la OMS; 4) el innovador importado de un país perteneciente al Consejo Internacional de Armonización (ICH, por sus siglas en inglés) o país observador ICH; y 5) el producto líder en el mercado 4. La normativa hace hincapié en el uso de un producto de referencia registrado y comercializado en el país; sin embargo, si el comparador no está disponible, DIGEMID permite adquirirlo de la lista de la OMS o proveniente de un país ICH 4. Para facilitar su identificación, el listado de productos de referencia para medicamentos en exigencia (obligatorio) 17 y para solicitudes voluntarias 18 se encuentran disponibles en el sitio web de la autoridad reguladora.
El medicamento de prueba debe presentar el mismo ingrediente farmacéutico activo (IFA), en la misma cantidad molar y forma farmacéutica, para la misma vía de administración que el IFA elegido como referencia, es decir, debe ser equivalente farmacéutico y, además, cumplir con los respectivos estándares de calidad. De esta manera, la equivalencia farmacéutica representa un requisito importante, que deberá realizarse con muestras del mismo lote 19. La tabla 1 presenta los requisitos que deben cumplir los medicamentos de prueba antes de someterse a los estudios de equivalencia terapéutica.
Tabla 1 Requisitos que deben cumplir los medicamentos de prueba antes de someterse a los estudios de equivalencia terapéutica.

IFA: ingrediente farmacéutico activo. Fuente: Decreto Supremo 024-2018-SA.
a Obligatorio en estudios de bioequivalencia.
Es importante destacar que los cambios significativos en las características fisicoquímicas del fármaco, en los excipientes y en el proceso de manufactura pueden afectar la biodisponibilidad del fármaco en el medicamento genérico, comprometer su equivalencia terapéutica y, en consecuencia, su intercambiabilidad 19 , 20. Tal es el caso de las formas farmacéuticas sólidas orales de liberación inmediata o modificada 19.
Otra consideración para garantizar la calidad de los medicamentos genéricos es el cumplimiento de las buenas prácticas de manufactura (BPM) que son, a la fecha, una de las exigencias establecidas por la DIGEMID para el otorgamiento del registro sanitario 21. Además, se apoya en las inspecciones periódicas de las plantas de producción, en la validación de los procesos productivos y en el cumplimiento de los requisitos de calidad según las farmacopeas oficiales, para garantizar que el producto no diferirá entre lotes y mantendrá la misma calidad que cuando se registró como genérico 22.
Cuando se llevan a cabo estudios in vivo, se debe garantizar el cumplimiento de las buenas prácticas clínicas (BPC) 13. Para tal efecto, se ha establecido que los estudios realizados en el Perú se ejecuten siguiendo el reglamento de ensayos clínicos del Instituto Nacional de Salud, y los realizados en el extranjero, según las guías de BPC de la OMS, ICH o las establecidas en el Documento de las Américas de la Conferencia Panamericana para la Armonización de la Reglamentación Farmacéutica y según los principios éticos de la Declaración de Helsinki 4. En la tabla 2 se enumeran las situaciones en las que es necesario el estudio de equivalencia terapéutica.
Tabla 2 Situaciones en las que es necesario el estudio de equivalencia terapéutica.
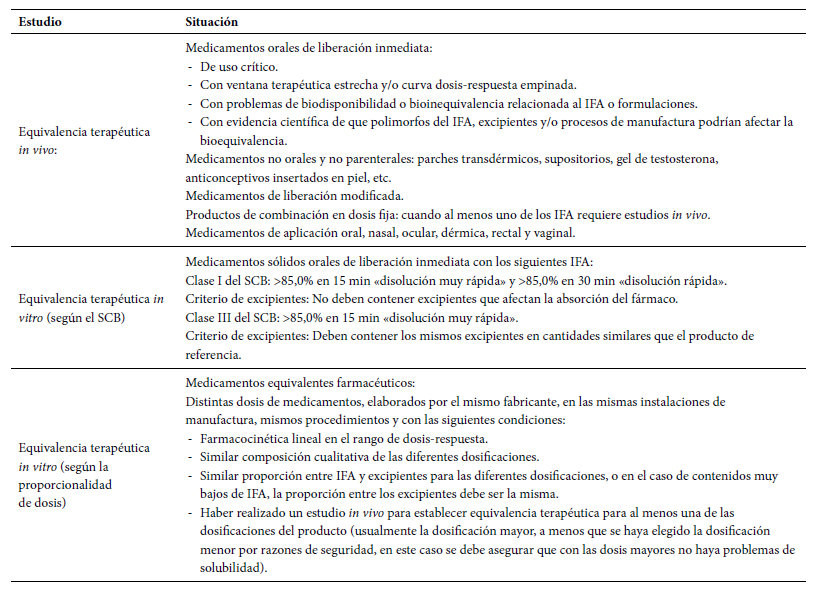
IFA: ingrediente farmacéutico activo; SCB: Sistema de Clasificación Biofarmacéutica. Fuente: Decreto Supremo 024-2018-SA
Por otro lado, existen formas farmacéuticas que no requieren estudios de equivalencia terapéutica. Se prescinde de estos estudios cuando las formas farmacéuticas presentan la misma concentración molar del IFA, con excipientes similares y de la misma función, de tal manera que no afecte la absorción del fármaco en comparación con el medicamento de referencia. En tales casos, la equivalencia farmacéutica, la comparación de las formulaciones, el cumplimiento de las BPM y la validación de los procesos productivos sustentan la intercambiabilidad del medicamento 13. Las formas farmacéuticas candidatas son las soluciones acuosas de uso parenteral, de uso oral (jarabes, elíxires y tinturas), de uso ótico u oftálmico, de uso tópico, para inhalación (nebulizadores o gotas nasales), los polvos para reconstitución (soluciones) y los gases. Estas tendrán un plazo de cinco años para demostrar que son intercambiables 4.
Además, en el caso de medicamentos reconocidos como intercambiables que posteriormente sufran cambios en su formulación en el lugar o proceso de fabricación, deberán presentar estudios de equivalencia terapéutica para demostrar la intercambiabilidad 4 , 14.
ESTUDIOS DE BIOEQUIVALENCIA Y BIOEXENCIÓN
La bioequivalencia es la comparación de las biodisponibilidades de dos productos (prueba y referencia) en términos de velocidad (representados por la concentración máxima, Cmáx, y el tiempo al que se alcanza esa concentración máxima, Tmáx) y el grado de absorción (representado por el área bajo la curva, ABC), después de la administración de la misma dosis molar y en las mismas condiciones, de tal manera que se espera que sus efectos clínicos sean similares 13.
El diseño de estudio convencional es cruzado, aleatorio, de dos períodos, dos secuencias y de dosis única y se realiza en voluntarios sanos bajo condiciones de ayuno 15 , 16. Asimismo, consta de tres etapas: clínica (administración de medicamentos de prueba y referencia en diferentes períodos y recolección de muestras en voluntarios), analítica (cuantificación del fármaco en muestras biológicas) y estadística 13.
Dos productos son bioequivalentes si los intervalos de confianza del 90% para el ABC y Cmáx del producto de prueba con respecto al comparador se encuentran entre el 80% y 125%. Para los productos terapéuticos de índice estrecho, se recomienda ajustar los intervalos de aceptación del ABC, y cuando la Cmáx sea de particular importancia para la eficacia, la seguridad o el control del nivel del fármaco, este rango de aceptación también debe ajustarse. El uso del Tmáx es necesario solo si existe relevancia clínica con respecto al momento de inicio de la acción o evidencia de efectos adversos 4 , 14.
No todos los medicamentos requieren estudios de bioequivalencia, existe la posibilidad de realizar estudios in vitro (o bioexención) para demostrar la intercambiabilidad, bajo ciertas consideraciones 23. Esta opción permite reducir el número de estudios en humanos, es menos costosa, requiere menos tiempo para su desarrollo 3 , 14 y asegura un bajo riesgo de bioinequivalencia cuando ocurren cambios después de su aprobación 24.
La bioexención se demuestra teniendo en cuenta el Sistema de Clasificación Biofarmacéutica (SCB) y la proporcionalidad de dosis ( 13. El SCB propuesto por Amidon et al. 25 categoriza los fármacos por su solubilidad acuosa y permeabilidad intestinal en altamente solubles y permeables (clase I), de baja solubilidad y muy permeables (clase II), muy solubles y de baja permeabilidad (clase III) y de baja solubilidad y permeabilidad (clase IV) y los productos farmacéuticos como «disolución rápida» y «disolución muy rápida» 25. Usando este enfoque, los IFA de clases I y III podrían ser candidatos a la bioexención; esta posibilidad solo es válida en fármacos de amplio margen de seguridad, en formas farmacéuticas sólidas de liberación inmediata de uso oral con excipientes ya aprobados y conocidos 13. Sin embargo, no se aplica en fármacos con un índice terapéutico estrecho ni tampoco en medicamentos de absorción bucal o sublingual 23. En todos los casos, debe demostrarse que los excipientes incluidos en la formulación están bien establecidos y no darán lugar a diferencias entre el producto de prueba y referencia en cuanto a la biodisponibilidad del fármaco 4.
La bioexención según el SCB se basa principalmente en una comparación de los perfiles de disolución entre los productos de prueba y referencia en medios de disolución de pH 1,2; 4,5 y 6,8. En todos los casos se deben cumplir los criterios de similitud (f2 entre 50 y 100) 13. La aplicación de la bioexención basada en la proporcionalidad de dosis de diferentes concentraciones de un producto farmacéutico solo es factible si las formulaciones tienen composiciones proporcionalmente similares 13 y bajo ciertas condiciones presentadas en la Tabla 2.
Con la nueva directiva se ha publicado la primera lista de medicamentos que incluye cinco IFA (ciclosporina, micofenolato de sodio, lamivudina, zidovudina y diazepam), que deberán demostrar equivalencia terapéutica con el medicamento de referencia, en plazos de presentación de tres años y dos años para los estudios in vivo e in vitro, respectivamente (Tabla 3) 4.
Tabla 3 Ingredientes farmacéuticos activos de medicamentos que deberán demostrar equivalencia terapéutica.
| IFA | Forma farmacéutica | Método |
|---|---|---|
| Ciclosporina | Cápsula blanda, solución oral | Bioequivalencia |
| Micofenolato de sodio | Tableta de liberación retardada | Bioequivalencia |
| Lamivudina | Tableta | Bioexención, SCB |
| Zidovudina | Tableta, cápsula | Bioexención, SCB |
| Lamivudina + Zidovudina | Tableta | Bioexención, SCB |
| Diazepam | Tableta | Bioexención, SCB |
| Topiramato | Tableta recubierta | Bioexención, SCB |
| Verapamilo (clorhidrato) | Tableta recubierta | Bioequivalencia |
| Warfarina (sódica) | Tableta | Bioequivalencia |
| Ácido valproico (valproato semisódico, divalproato sódico) | Tableta de liberación prolongada | Bioequivalencia |
| Ácido valproico (valproato semisódico, divalproato sódico) | Tableta de liberación retardada, comprimido recubierto gastrorresistente, tableta con recubierta entérica | Bioequivalencia |
| Valproato sódico | Tableta de liberación retardada, comprimido recubierto gastrorresistente, tableta con recubierta entérica | Bioequivalencia |
| Fenitoína (sódica) | Cápsula | Bioequivalencia |
| Lamotrigina | Tableta, comprimido, tableta dispersable | Bioequivalencia |
| Levetiracetam | Comprimido recubierto | Bioexención, SCB |
| Levetiracetam | Tableta de liberación prolongada | Bioequivalencia |
| Tacrolimus | Cápsula | Bioequivalencia |
| Teofilina | Tableta de liberación sostenida | Bioequivalencia |
| Levodopa + carbidopa | Tableta | Bioexención, SCB |
| Levotiroxina sódica | Tableta | Bioequivalencia |
| Oxcarbazepina | Tableta | Bioequivalencia |
| Micofenolato de mofetilo | Cápsula, tableta recubierta | Bioequivalencia |
| Azatioprina | Comprimido recubierto | Bioequivalencia |
| Carbamazepina | Tableta | Bioequivalencia |
| Carbonato de litio | Tableta | Bioequivalencia |
| Digoxina | Tableta | Bioequivalencia |
IFA: ingrediente farmacéutico activo; SCB: Sistema de Clasificación Biofarmacéutica. Fuente: Decreto Supremo 024-2018-SA y Resolución Ministerial 404-2021/MINSA.
PERSPECTIVAS FUTURAS
En países desarrollados como los Estados Unidos y los de la Unión Europea, la equivalencia terapéutica es un requisito reglamentario para el registro de medicamentos genéricos. En América Latina, este requisito difiere considerablemente, a pesar de los esfuerzos de armonización 26. Países como México, Brasil y Argentina comercializan genéricos intercambiables. Por el contrario, en países como Bolivia, Honduras, Nicaragua, República Dominicana, entre otros, la comercialización de los productos farmacéuticos sin pruebas de equivalencia terapéutica sigue siendo una realidad actual 27.
En el Perú, se adoptó el nuevo requisito de intercambiabilidad para el registro sanitario de productos farmacéuticos multifuentes con el respaldo de estudios de equivalencia terapéutica, cuya identificación se consignará en el rotulado del empaque externo con una banda verde con el nombre «Medicamento intercambiable» 18. Esta implementación contribuirá a recuperar la confianza en los medicamentos genéricos y fomentará un entorno donde médicos, farmacéuticos y consumidores utilicen alternativas genéricas disponibles, en vista de su intercambiabilidad con el medicamento de referencia 28.
A dos años de la aprobación del reglamento, se han publicado los primeros medicamentos que han demostrado equivalencia terapéutica 18 y el segundo listado donde se añadieron diecinueve IFA al listado inicial (Tabla 3) 29.
Se espera que, dentro de los plazos establecidos, los medicamentos en exigencia presenten los resultados de los estudios in vivo e in vitro con el medicamento de referencia y, en general, los titulares de los registros sanitarios presenten el requisito de intercambiabilidad en el procedimiento de inscripción y reinscripción de sus productos farmacéuticos. Mientras tanto, los medicamentos multifuentes continúan comercializándose ante la ausencia de estudios de equivalencia terapéutica. Hallazgos encontrados demuestran que algunos productos farmacéuticos fallaron en las comparaciones in vivo e vitro con el medicamento de referencia (Tabla 4) 30 - 40, representando un riesgo potencial para la salud de la población.
Tabla 4 Hallazgos de investigaciones realizadas en productos farmacéuticos multifuentes.
| IFA | Método | Hallazgos | Cita |
|---|---|---|---|
| Ibuprofeno | in vivo | PM es bioequivalente al PR | 30 |
| Diclofenaco | in vivo | PM no es bioequivalente al PR | 31 |
| Amoxicilina | in vitro | Solo 2 de 3 PM son equivalentes al PR | 32 |
| Metronidazol | in vitro | Ningún PM es equivalente al PR | 32 |
| Diazepam | in vitro | Solo 2 de 3 PM son equivalentes al PR | 33 |
| Paracetamol, clorfenamina maleato, fenilefrina clorhidrato | in vitro | Solo 2 de 4 PM son equivalentes al PR | 34 |
| Metformina clorhidrato | in vitro | Ningún PM es equivalente al PR | 35 |
| Isoniazida | in vitro | Solo 1 de 2 PM es equivalente al PR | 36 |
| Amoxicilina | in vitro | Solo 2 de 4 PM son equivalentes al PR | 37 |
| Doxiciclina | in vitro | Todos los PM son equivalentes al PR | 37 |
| Fluconazol | in vitro | Ningún PM es equivalente cal PR | 37 |
| Rifampicina | in vitro | Ningún PM es equivalente al PR | 38 |
| Fenitoina | in vitro | Todos los PM son equivalente al PR (a pH 1,2) | 39 |
| Glibenclamida | in vitro | Todos los PM son equivalente al PR (a pH 6,8) | 40 |
IFA: ingrediente farmacéutico activo; PM: producto farmacéutico multifuente; PR: producto de referencia.
Fuente: Elaboración propia.
La exigencia de la intercambiabilidad de medicamentos en el Perú representa un gran desafío para los laboratorios fabricantes. La capacidad de los fabricantes locales para realizar estudios in vivo, la acreditación de los centros de investigación, el acceso limitado al medicamento de referencia y los estrictos plazos establecidos representan algunos de los desafíos por superar si se desean incrementar el registro sanitario de medicamentos genéricos intercambiables 41.
CONCLUSIONES
Implementar la intercambiabilidad de medicamentos en el Perú representa un esfuerzo del Ministerio de Salud para reducir los costos de las terapias farmacológicas, a fin de que la población tenga un mayor acceso a medicamentos genéricos (multifuentes) eficaces, seguros y de calidad. Mientras tanto, se siguen comercializando medicamentos en ausencia de estudios de equivalencia terapéutica. Los estudios demuestran que algunos medicamentos fallaron en las comparaciones in vivo e in vitro con el medicamento de referencia. La nueva exigencia representa un gran desafío para los laboratorios fabricantes y los titulares de registros sanitarios a fin de demostrar la intercambiabilidad de sus productos farmacéuticos con el medicamento de referencia.

