











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El término de error innato del metabolismo (EIM) fue introducido en el año 1909 por Sir Archibald Garrod, quien describió a la alcaptonuria, albinismo cistinuria y pentosuria; incorporando la idea de la «individualidad bioquímica» 1. Los EIM son un grupo de enfermedades que tienen una prevalencia de 1 por cada 1000 personas 2. El número de EIM conocidos a la fecha son aproximadamente 1015 entidades bien definidas, dentro de las cuales se encuentran los trastornos intracelulares de la cobalamina 3.
La cobalamina es convertida en dos coenzimas: metilcobalamina (MeCbl) y adenosilcobalamina (AdoCbl). La MeCbl es utilizada para la conversión de homocisteína a metionina, a través de la metionina sintasa; mientras que la AdoCbl es utilizada por la enzima mitocondrial metilmalonil CoA mutasa, la cual convierte a la L-metilmalonil-CoA a succinil-CoA 4. Los trastornos intracelulares de la cobalamina (TIC) son clasificados en nueve grupos de complementación genética (cblA-G, J y X) 5. La aciduria metilmalónica y homocistinuria tipo cblC o ACMMHC (MIM #277400) es el trastorno más común dentro de los TIC, el cual es provocado por variantes en el gen MMACHC, localizado en 1p34.14, causando una disminución de AdoCbl, ni MeCbl, lo que se manifiesta clínicamente por dificultades en la alimentación, fallo de medro, trastornos hematológicos, neurológicos, renales, oftalmológicos y dermatológicos 6. Otras manifestaciones son hidrops fetalis7, retraso del crecimiento intrauterino, anomalías cardíacas 8, cardiomiopatía dilatada 9, esteatosis hepática, enteropatía perdedora de proteínas y linfohistiocitosis hemafagocítica 10. Dentro de las manifestaciones bioquímicas son vitamina B12 dentro de valores normales, homocistinuria, homocistinemia, aciduria metilmalónica, metionina disminuida, cistationemia y rara vez hiperamonemia con acidosis metabólica 7.
La enfermedad por cblC se clasifica según las características clínicas en fenotipo severo y moderado. El primero con manifestaciones sistémicas desde el primer año de vida (neurológicas, hematológicas y oftalmológicas) y una mortalidad de la cuarta parte; y el segundo con manifestaciones extrapiramidales, demencia, delirio o psicosis 11. El tratamiento en ambos grupos es con hidroxicobalamina, betaína, ácido fólico y carnitina 10,11. Algunos pacientes, a pesar del tratamiento, podrían desarrollar severas complicaciones; más aún si el tratamiento no fue oportuno 10. La efectividad de la cobalamina parenteral se ve respaldada por la mayor supervivencia de los pacientes que desarrollan el síndrome urémico-hemolítico 12 y la reversión de complicaciones graves 10. El uso de dosis más altas de cobalamina parenteral ha sido eficaz para reducir los metabolitos y ha llevado a la resolución de la microangiopatía trombótica en pacientes con enfermedad por cblC 13. La incidencia de la enfermedad por cblC es variable según las poblaciones y en Perú se desconoce la incidencia de esta entidad 10.
Se describe el primer paciente por deficiencia de cblC en Perú, haciendo hincapié en la historia natural de la enfermedad hasta los dos años y seis meses de edad; y luego como muchos de los síntomas y signos se revierten progresivamente, asimismo, se plantea la importancia del tamizaje neonatal ampliado.
REPORTE DE CASO
Paciente varón de dos años seis meses, nacido de parto vaginal a término, con peso al nacer de 2735 kg, talla de 50 cm, perímetro cefálico desconcido y con puntaje de Apgar de 81-95. Tiene una hermana mayor sin antecedentes médicos y los padres son consanguíneos (F=1/8) (Figura 1A). La antropometría para la talla, perímetro cefálico y peso se encuentra en los percentiles de 57, 13 y 15 respectivamente; su desarrollo psicomotor tuvo un retraso, observándose que el control cefálico fue al año, control torácico con apoyo al año seis meses, y sonrisa social al año un mes. Al examen físico no se observan dismorfias, pero presenta pelo ralo, livedo reticularis, tono muscular disminuido a predominio axial.
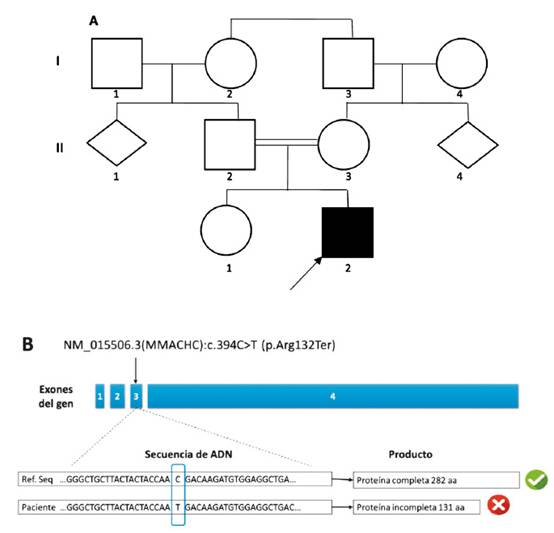
Figura 1 A. Árbol genealógico, nótese que los padres (II.2 y II.3) del propositus (III.1) tienen un coeficiente de endogamia (F) de 1/8. B. Impacto clínico de la variante patogénica del gen MMACHC. Ubicación de la variante patogénica c.394C>T (p.Arg132Ter) en el gen MMACHC y la consecuente producción de una proteína truncada con alteración de función; en azul: exones del gen MMACHC (secuencia de referencia del ADN normal).
Presentó varias hospitalizaciones, al nacer por ictericia, a los 13 días de vida por neutropenia severa, a los siete meses por otitis media aguda y neutropenia; 11 meses por epilepsia; un año dos meses por epilepsia, pancitopenia, impétigo, estomatitis aftosa. Para cada episodio de neutropenia recibió filgastrin, no observándose una respuesta favorable.
Los exámenes complementarios a los 11 meses de edad se observan en la Tabla 1. Por los resultados alterados, se realizó la determinación de variantes patogénicas de la acidemia propiónica (E168K, 1218del, 14/ins 12, 1170 insT) y de la acidemia metilmalónica (N129Y, G717V), no observándose cambios. Los exámenes que estuvieron dentro de la normalidad fueron los potenciales evocados auditivos, electroencefalograma, perfil tiroideo, cariotipo 46,XY; citometría de flujo de CD 19+, CD3+, CD4+, CD8+, CD16/56+, ecografía renal y hepática. En la resonancia magnética de encéfalo se observa hipotrofia cerebral de lóbulos frontales y temporales, retraso de la mielinización de la sustancia blanca.
Tabla 1 Principales resultados de laboratorio basal y postratamiento.
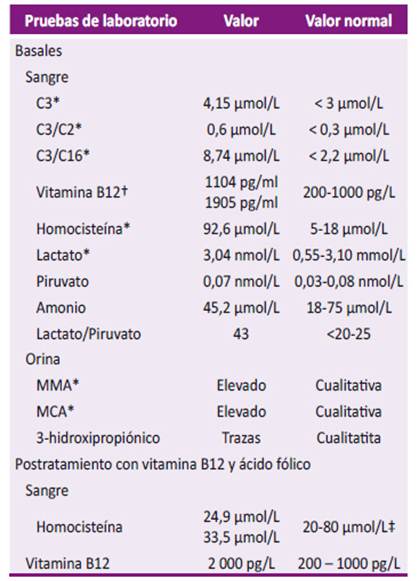
C2: acetil-carnitina; C3: propionil-carnitina; C16: palmitoil-carnitina; C3/C2: propionil-carnitina/acetil-carnitina; C3/C16: propionil-carnitina/palmitoil-carnitina; MMA: ácido metilmalónico; MCA: ácido metilcítrico.
* Con correlación bioquímica según el fenotipo cblC. † No existe correlación bioquímica con fenotipo cblC. ‡ Valores normales en pacientes tratados.
El secuenciamiento exómico clínico encontró una variante homocigota en el gen MMACHC en c.394 C>T, el cual provoca un codón de parada (Figura 1B), por lo que fue clasificada como patogénica y está disponible en la base de datos ClinVar (VCV000001423.6) con la nomenclatura NM_015506.3(MMACHC):c.394C>T (p.Arg132Ter); no se encontró otras variantes patogénicas incidentales.
Se inició tratamiento al año cinco meses con hidroxicobalamina a dosis de 1 mg intramuscular diario y de ácido fólico de 0,5 mg diarios. Un control a los cuatro meses de iniciado el tratamiento, se observó una disminución en plasma de homocisteína y persistencia de vitamina B12 alta (Tabla 1). En el último control a los dos años y tres meses, el hemograma estaba dentro de parámetros normales.
DISCUSIÓN
El diagnóstico inoportuno en este paciente hizo que las complicaciones neurológicas y hematológicas sean más graves, manifestadas como un retraso del desarrollo psicomotor severo, además de neutropenias recurrentes provocando múltiples hospitalizaciones. Además, nos mostró que las manifestaciones bioquímicas fueron diferentes con relación a los valores de vitamina B12, donde se describe que en la mayoría de los pacientes se encuentra normal. Planteamos que esta variante bioquímica se debe a que la mayoría de los niños descritos previamente se diagnostican en la etapa neonatal, y que la demora en el diagnóstico hizo que progresivamente los valores de esta vitamina aumenten notoriamente. Por la falta de una correlación bioquímica con algún error innato del metabolismo de la cobalamina, hizo que se planteara como parte de la terapéutica la inclusión de fórmulas especiales para la aciduria metilmalónica, y por las neutropenias se planteó el diagnóstico de inmunodeficiencia primaria. El diagnóstico de los TIC se basa utilizando la cromatografía de gases acoplada a espectrometría de masas en tándem o GC-MS 14 de ácido metilmalónico en orina y plasma, así como de homocisteína; seguido de la determinación de las variantes patogénicas bialélicas de los diferentes grupos de complementación 7. La GC-MS necesita de muestras congeladas, lo que hace complicada la conservación y el envío de la muestra. Sin embargo, lo que nos definió el diagnóstico fue el procesamiento de una muestra de sangre seca en papel filtro utilizando el secuenciamiento exómico clínico, facilitado por una mejor conservación y fácil envío. La respuesta bioquímica de la homocisteína al tratamiento estuvo dentro de lo esperado (20-80 µmol/L) 7.
El tamizaje neonatal son programas de salud pública utilizadas mundialmente hace más de 50 años (estrategias de prevención secundaria), que se realizan a los recién nacidos asintomáticos y que no tienen un riesgo incrementado de algún problema de salud 15. El principal objetivo es prevenir o mejorar a largo plazo consecuencias de alguna enfermedad potencialmente tratable 15. En Perú, se tiene normado desde el 2019, el tamizaje neonatal de hipotiroidismo congénito, hiperplasia suprarrenal congénita, fenilcetonuria, fibrosis quística, hipoacusia congénita y catarata congénita 16. Mientras que, en Latinoamérica el tamizaje neonatal se inicia desde los años 90s, llegando a tener países que realizan el tamizaje de hasta 29 condiciones (Tabla 2) 17,18.
Tabla 2 Panorama actual del tamizaje neonatal universal en países de Latinoamérica.
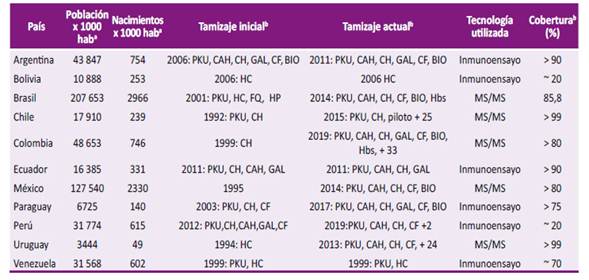
a Datos obtenidos de UNICEF. El Estado Mundial de la Infancia 2017: Niños en un mundo digital. New York: UNICEF; 2017
b Adaptado de Therrell et al. Current status of newborn screening worldwide: 2015. Program demographics and screened conditions in Latin American screening programs; y UNICEF. El Estado Mundial de la Infancia 2017: Niños en un mundo digital. New York: UNICEF; 2017
PKU: fenilcetonuria; HAC: hiperplasia adrenal congénita; CH: hipotiroidismo congénito; CF: fibrosis quística; GAL: galactosemia; BIO: déficit biotinasa; Hbs: hemoglobinopatías; MS/MS: espectrometría de masas en tándem.
El presente caso nos muestra la importancia del tamizaje neonatal en las enfermedades potencialmente tratables con tecnologías más costo/efectivas que las utilizadas actualmente, como la espectrometría de masas en tándem 19. Inclusive se han reportado resultados favorables por la metodología de secuenciamiento del gen MMACHC en muestras de vellosidades coriónicas de gestantes como parte del diagnóstico genético prenatal 20,21.
El diagnóstico bioquímico de las acidurias orgánicas, en la mayoría de los casos se debe realizar a través de tecnologías como la cromatografía de masas en tándem en muestras de sangre y orina. Este tipo de pruebas no se encuentran disponible en nuestro medio, por lo que es de suma importancia tener convenios directos con laboratorios internacionales o mejor aún, implementar estas técnicas en al menos un centro de referencia, lo cual disminuiría notablemente los costos en el diagnóstico repercutiendo de manera directa en un manejo más efectivo. En tal sentido, las políticas públicas deberian establecer la prevalencia de estas entidades en estudios pilotos y según los resultados realizar su priorización en base a criterios ya establecidos desde hace mucho tiempo; no sólo con la intención de poder ofrecer tratamientos dirigidos, sino también con la finalidad de poder indicar el riesgo de recurrencia familiar y disminuir mediante tecnologías de reproducción asistida el número de niños con patologías devastadoras.

