











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La ocratoxina A es comúnmente producida por los mohos Aspergillus ochraceus y Penicillium viridicatum; su presencia está asociada a los productos como el maíz, la cebada, el café verde y varias frutas secas1. La ocratoxina A puede estar presente en conjunción con las aflatoxinas, sustancias consideradas como carcinógenos naturales. La ocratoxina A es un potencial agente carcinógeno, que afecta los riñones de los animales expuestos a dicha micotoxina2.
Se estima que una cuarta parte de las cosechas mundiales están contaminadas por micotoxinas. Un estudio analizó 2 753 muestras de cereales y piensos, revelando que más de la mitad de las muestras de origen europeo y la tercera parte de la región Asia - Pacífico estaban contaminadas con niveles de micotoxinas por encima del límite de cuantificación3.
El consumo mundial de café supera los seis millones de toneladas al año y la venta minorista, principalmente en Europa, EE.UU. y el Japón, se encuentra alrededor de los 70 000 millones de dólares al año. Los países en desarrollo que producen la materia prima reciben unos 6 000 millones de dólares 4.
Aunque no hay un nivel consultivo o regulatorio para la ocratoxina A emitido por la Administración de Alimentos de los EE.UU., muchos coinciden que los niveles entre 10-20 partes por mil millones (ppb) en productos destinados al consumo humano o animal pueden causar problemas de salud y pérdidas económicas. Algunos mercados extranjeros han establecido límites de regulación que van desde 5-50 ppb para el comercio interno, mientras que la Comisión Europea desde el año 2006 ha establecido un límite máximo de 5 ppb para café tostado5.
La mejor protección contra las micotoxinas es controlar su presencia en piensos y alimentos. Eso significa mantener un control en todo el camino productivo desde la cosecha inicial de granos hasta el producto terminado.
Parte experimental
El estudio se realizó en el Instituto de Investigación en Ciencias Farmacéuticas y Recursos Naturales “Juan de Dios Guevara” de la Facultad de Farmacia y Bioquímica de la Universidad Nacional Mayor de San Marcos y en el Laboratorio de Investigación y Certificaciones (LABICER) de la Facultad de Ciencias de la Universidad Nacional de Ingeniería.
Reactivos
Acetonitrilo de grado HPLC de la marca J.T Baker, estándar de ocratoxina A (OTA) 99,18 %, metanol grado HPLC (Merck), ácido acético (CH3COOH) grado analítico 100 %, Buffer fosfato salino en tabletas pH=7,40 (PBS) y Tween 20 adquiridos de la casa comercial Merck. El agua utilizada fue purificada en un equipo Elga Purelab CLASSIC UV.
Equipos
Cromatógrafo Líquido de Alto Rendimiento-SHIMADZU que posee: desgasificador DGU- 20A5R, automuestreador SIL-30AC, bomba cuaternaria LC-30AD, horno de columna: CTO-20AC, detector de fluorescencia RF-20Axs, control de sistema CBM-20A; Columna cromatográfica RP C18 de 250 mm x 4,6 mm ID x 5 µm partícula (Restek); Molinillo Bosch, agitadores magnéticos de las marcas Thermo Scientific Cimarec e IKA C-MAG HS7; Balanza analítica digital GR-300 AND, equipo ultrasonido WISD Laboratory Instruments; Ultrapurificador de agua ELGA PURELAB CLASSIC UV y columnas de inmunoafinidad para ocratoxina NEOCOLUMN.
Colecta de la muestra
Los granos de café tostado provinieron del departamento de Junín, provincia de Chanchamayo, distrito Perené del anexo Pampa Tigre. Las actividades de muestreo se realizaron a temperatura ambiente y humedad atmosférica característica de la zona en el mes de noviembre de 2018. Las muestras fueron rotuladas y transportadas al departamento de Lima para su tratamiento y respectivos análisis.
Tratamiento de la muestra
Los granos de café fueron secados en una estufa a 38°C por 24 horas. Las muestras de café tostado se molieron en un molinillo Bosch. Una vez trituradas se homogenizaron y se conservaron a temperatura de refrigeración en el rango de 0°C a 4°C para su posterior análisis.
Preparación de los estándares de ocratoxina A
La solución principal del estándar de OTA se preparó diluyendo 1 mg de estándar OTA en 50 mL de mMetanol, teniendo una concentración de 20 ppm. A partir de la solución principal, se preparó la solución de trabajo a una concentración de 200 ppb y a partir de esta solución se prepararon los diferentes estándares utilizados para la construcción de la curva de calibración, cuyas concentraciones variaron de 0,05 a 10 ppb. De la solución madre se prepararon las soluciones utilizadas en las pruebas de recuperación del método utilizado en granos tostados.
Extracción y purificación de la ocratoxina A en las muestras
Se utilizó 10 g de café tostado finamente molido, y pasado por un tamiz de 0,5 mm. Para las extracciones se utilizó 100 ml de metanol con bicarbonato en una proporción de 7:3 de metanol y solución de bicarbonato de sodio al 1 %, respectivamente. Luego, se mezcló en un agitador magnético a alta velocidad durante tres minutos, se dejó en reposo por tres minutos y se filtró el sobrenadante líquido con un papel Whatman N°426-7.
Se mide cinco mililitros del filtrado y se diluye con 45 ml de PBS / Tween (0,01% v / v) obteniendo un volumen final de 50 ml. Con una de jeringa de 50 ml y un adaptador de columna, se eluyó la muestra de 50 ml a través de la columna de inmunoafinidad gota a gota. Se tomó en consideración que la columna no se seque. Posteriormente, se lavó la columna con 20 ml de PBS / Tween (0,01% v / v) y con 10 ml de PBS, manteniendo las mismas consideraciones. Se aseguró en eliminar todo el líquido de la columna utilizando presión positiva de una jeringa8. La OTA se reconstitutyó con 0,75 mL de metanol acidificado; 0,75 mL de metanol y 1,5 de agua ultrapura, según las indicaciones del fabricante de Columnas de Inmunoafinidad Neocolumn.
Cuantificación por cromatografía líquida de alto rendimiento
Los análisis se realizaron mediante cromatografía líquida de alto rendimiento con detección por fluorescencia, la metodología y las condiciones del equipo propuestas por Tafuri9.
La longitud de onda de excitación fue de 333 nm y la longitud de onda de emisión fue de 460 nm. La temperatura del horno de la columna fue de 25 °C. La elución fue a un flujo constante de 1 mL/min de CH3CN (ácido acético 1%) y H2O (ácido acético 1 %) (50:50 v/v) como el sistema inicial de eluyentes. La gradiente de elución, se inició con el acondicionmiento del sistema de solventes por 20 min desde 50 % hasta 100 % de CH3CN la cual se mantuvo durante 3 minutos; los últimos 10 minutos el sistema se restableció a sus condiciones iniciales de CH3CN 50 % y H2O 50 %.
Los eluyentes y las muestras fueron filtrados a través de filtro de jeringa (0,22 µm), el volumen de inyección a la columna cromatográfica fue de 25µL.
Las lecturas de los estándares de OTA y muestras problemas se realizaron por duplicado. La identificación de OTA se realizó utilizando el tiempo promedio de retención de las diluciones de los estandares. Este tiempo de retención y Límite de Detección (LDD) se determinó midiendo diez veces el estándar de trabajo a una concentración de 0,09918 ppb, El tiempo de retención promedio fue de 7,964 ± 0,010 minutos y el LDD promedio fue de 0,019 ± 0.001 ppb.
Pruebas de recuperación del método utilizado
Para determinar la eficacia en el proceso de recuperado se procedió elaborando un blanco para la muestra de granos de café tostado, adicionado a una concentracion de 5 ppb con estándar de OTA. Las muestras adicionadas se trataron de igual manera que las muestras problemas.
Resultados y discusión
La identificación de la ocratoxina A se realizó utilizando el tiempo de retención promedio de las diluciones de trabajo, el cual fue de 7,964 min, a partir del estándar de menor concentración (0,09918 μg /L) se realizaron 10 repeticiones.
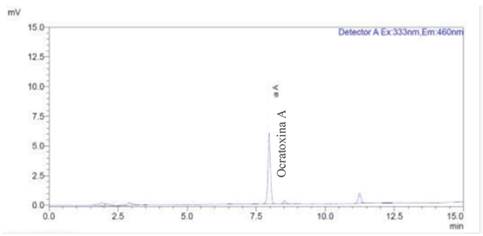
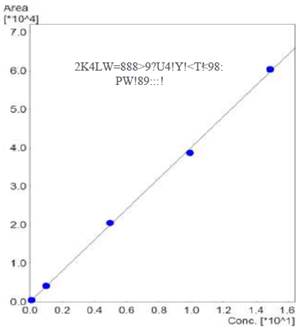
Figura 2 Curva de calibración utilizada para la cuantificación de ocratoxina A a concentraciones promedios de 0,099; 0,991; 4,959; 9,918; 14,877 μg/L.
El método de recuperación utilizado para el análisis de OTA fue evaluado al nivel de 5 ppb (a) de OTA, los resultados se muestran en la tabla 1.5
Tabla 1 Contenido de OTA en las muestras adicionadas de café tostado.
| Muestra | Tiempo de retención | Área | Altura | Concentración | %RSD* |
|---|---|---|---|---|---|
| Café tostado | 7,982 | 2697 | 416 | 0,643 μg/L | 1,760 |
| Muestra adicionada con OTA | 7,981 | 2634 | 415 | 0,627 μg/L | |
* RSD= desviación estándar relativa. El porcentaje de recperación fue de 76,231 % evaluando al nivel de 5 ppb
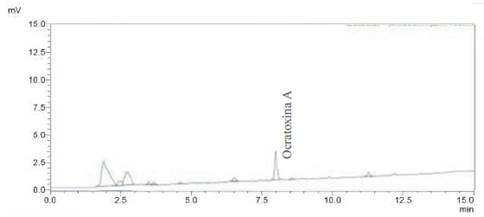
El propósito de la extracción es fijar la ocratoxina A en la matriz de análisis, es decir sobre el material de la columna de inmunoafinidad, usando solventes adecuados para su posterior recuperación y cuantificación. Los solventes de extracción y el método utilizado son las dos consideraciones más importantes para el procedimiento de extracción de ocratoxina A10. La selección del solvente de extracción depende de varios factores, incluidas las características físicas y químicas del analito, el costo y la seguridad del solvente, la solubilidad de compuestos diferentes a la ocratoxina A (interferencia por colorantes u otros componentes) y los procesos después de la extracción11.
En la investigación se utilizó columnas de inmunoafinidad para ocratoxina A de la marca NeoColumn código del producto 8640, fabricadas por NeoGen® Corporation, el cual posee un método de extracción validado para muestras de café verde, tostado e instantáneo, aunque existe diversos variantes de los sistemas de solventes de extracción de OTA en café9,10,11,12.
Se recomienda el uso de una disolución de metanol al 60 % y agua al 40 %12. También, se han utilizado como solvente de extracción metanol y solución de bicarbonato de sodio al 3 % en la relación 1:113. Al saber que la OTA es una molécula de polaridad media, se busca el uso de solventes polares y semipolares, esto es contrastable en diversas investigaciones, aunque suelen existir modificaciones para permitir mayores eficiencias en la extracción relacionadas a menos efectos de interferencia. Existen reportes utilizando solventes como la acetona, acetato de etilo, cloroformo para la extracción de ocratoxina A en matrices complejas14,15,16, cabe acotar que el uso de solventes apolares como en acetato de etilo para la extracción de OTA, puede ser necesario bajo un procedimiento de desengrase adicional antes de la detección o purificación, adicionalmente del uso de solventes semipolares acidificados para su reconocimiento por HPLC14.
Las muestras adicionadas con OTA presentaron un porcentaje de recuperación de 76,23% al nivel de 5 ppb. Los límites de recuperación comúnmente aceptados para un nivel de contaminación de OTA de 10 ppb están comprendidos entre 70 a 125 % de recuperación17.
Para la concentración evaluada a 5 ppb de OTA, el porcentaje de recuperación del método de la Comunidad Europea5, se encontró entre estos los límites. Sin embargo, existen métodos en donde el porcentaje de recuperación para concentraciones más bajas de OTA (5 ppb) fue inferior al 70 % con el método validado por la Unión Europea (UE) “14132”15. La recuperación por el método de Pittet18, el cual emplea solventes como metanol y bicarbonato de sodio al 3 %, difiere del método usado en los volúmenes de extracción y soluciones estabilizadas de lavado (PBS pH 7,4).
Las condiciones de los solventes que presenten un pH demasiado alto conducen a la formación de OTA de anillo abierto (OP-OA), que no es reconocido por los anticuerpos OTA en el procedimiento de purificación o limpieza. Estas circunstancias ya han sido reportadas durante el análisis de otras matrices contaminadas con OTA19,20.
La dilución realizada en nuestro método y el uso de PBS ajustado a 7,4 de pH, permite que se realice la conversión de OP-OTA en OTA antes del proceso de purificación por columnas de inmunoafinidad. Los valores de recuperación son relativamente constantes con un promedio del 70 %. Se recomienda que las recuperaciones inferiores al 60 % pueden estar sujetas a investigaciones que conduzcan a una mejora en los métodos y solventes de extracción17. Manteniendo la línea de investigación en la búsqueda de eficacia de los métodos de recuperación.
Los resultados obtenidos de OTA en muestras de granos de café tostado se muestran en la tabla 2.
Tabla 2 Contenido de OTA en las muestras de café tostado.
| Muestra | Tiempo de retención | Área | Altura | Concentración | %RSD |
|---|---|---|---|---|---|
| Café tostado | 8,077 | 407 | 37 | 0,071 μg/L | 1,499 |
| M1 | 8,066 | 413 | 37 | 0,073 μg/L | |
| Café tostado | 8,071 | 704 | 74 | 0,142 μg/L | 2,354 |
| M2 | 8,070 | 723 | 75 | 0,150 μg/L |
Las muestras fueron analizadas por duplicado reportando valores de 0,072 ± 0,001 y 1,248 ± 0,003 μg/L equivalentes a 0,216 y 0,444 ppb de OTA. Los resultados no superan los límites máximos permitidos de 5 ppb OTA en café tostado y molido, establecidos por la Unión Europea.
Tabla 3 Cálculo de Límite de Detección (LDD)
| Repetición | Tiempo de retención | Área | Altura | Concentración | Ruido | LLD* |
|---|---|---|---|---|---|---|
| 1 | 7,959 | 463 | 75 | 0,0853 μg/L | 5,28 | 0,020 |
| 2 | 7,955 | 443 | 73 | 0,0804 μg/L | 5,40 | 0,020 |
| 3 | 7,961 | 460 | 75 | 0,0845 μg/L | 5,35 | 0,020 |
| 4 | 7,963 | 456 | 73 | 0,0837 μg/L | 5,22 | 0,020 |
| 5 | 7,978 | 449 | 73 | 0,0819 μg/L | 4,81 | 0,018 |
| 6 | 7,971 | 448 | 73 | 0,0817 μg/L | 5,50 | 0,020 |
| 7 | 7,954 | 464 | 75 | 0,0857 μg/L | 5,10 | 0,019 |
| 8 | 7,954 | 471 | 75 | 0,0872 μg/L | 5,01 | 0,020 |
| 9 | 7,981 | 455 | 73 | 0,0834 μg/L | 4,60 | 0,017 |
| 10 | 7,989 | 448 | 71 | 0,0816 μg/L | 5,12 | 0,019 |
| Promedio | 7,964 | 456,6 | 73,9 | 0,0838 μg/L | 5,14 | 0,019 |
* Se calculó mediante la siguiente fórmula, LLD= 3,3 x (Concentración) x Ruido/altura del pico
CONCLUSIONES
El proceso de extracción, purificación y cuantificacion de ocratoxina A en Coffe arábiga L. “café”, de tipo tostado es aceptable aplicando el método de columna de inmunoafinidad y su detección por fluorescencia, corroborado el nivel concentración de 5 ppb. Las concentraciones de las muestras analizadas no superan el límite permitido por los organismos reguladores.

