











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Una molécula con potencial actividad farmacológica debe poseer ciertas características químicas que favorezcan su biodisponibilidad, permeabilidad celular, solubilidad, entre otros. Muchas propiedades químicas o descriptores se pueden estimar con modelos computacionales o in silico y pueden facilitar su selección en bases de datos moleculares, para luego ser estudiadas in vitro. Existen reglas empíricas para evaluar la capacidad de una molécula de convertirse en un fármaco, conocida como drogabilidad o druglikeness, y algunas de las reglas más populares son las de Lipinski (1), Ghose (2), Veber (3), Egan (4) y Muegge (5). Estas consideran un rango de valores para la masa molecular, el área de superficie polar topológica (TPSA), el número de átomos capaces de formar enlaces de hidrógeno, entre otros, como se muestra en la Tabla 1.
Tabla 1 Druglikeness de reglas empíricas clásicas.
| Modelo | Druglikeness | Ref. |
| Lipinski | MW<500, logP<4.15, N ó O <10, NH ó OH <5 | (1) |
| Ghose | 160<MW<480, -0.4<logP<5.6, 40<MR<130, 20<átomos<70 | (2) |
| Veber | Enlaces rotables <10, TPSA <140 | (3) |
| Egan | logP<5.88, TPSA<131.6 | (4) |
| Muegge | 200<MW<600, -2<logP<5, TPSA<150, anillos<7, C>4, heteroátomos>1, enlaces rotables< 15, HBA<10, HBD<5 | (5) |
MW: masa molecular, logP: coeficiente de partición agua-octanol, MR: refractividad molar, TPSA: área superficial polar topológica, HBA: aceptor de enlace puente de hidrógeno, HBD: donador de enlace puente de hidrógeno.
Uno de los parámetros más recurrentes es el coeficiente de partición, que consiste en la relación de las concentraciones de la molécula en estudio entre dos disolventes inmiscibles en equilibrio. El coeficiente de partición octanol-agua (logP) se utiliza como una medida de lipofilia y un indicador general de la permeabilidad celular (6), con valores óptimos reportados entre 1 y 3. Se han desarrollado varias herramientas para la predicción de logP (7), principalmente para ahorrar en reactivos químicos, esfuerzo y tiempo. De este modo, encontramos métodos basados en mecánica cuántica (ab initio, semiempíricos, DFT, etc.), que requieren el cálculo de la energía electrónica de la molécula en octanol, agua y vacío, para estimar el logP mediante la determinación del valor de la Energía Libre de Gibbs. Estos métodos son menos adecuados para su aplicación en grandes bases de datos debido a su alto costo computacional. Métodos menos costosos, y por lo tanto considerados estándar, se basan en la suma de las contribuciones al logP por átomo o grupo funcional (también llamado fragmento) (8,9,10,11,12,13,14). Aunque estos métodos presentan un buen grado de predicción, tienen diferentes limitaciones, como valores irreales, sesgo que subestima el logP, número limitado de átomos de metales de transición, entre otros. Con respecto al uso de metales de transición (15), es necesario generar bases de datos de compuestos bioinorgánicos (16) debido al significativo potencial farmacológico representado por complejos o compuestos de coordinación entre biometales y fármacos orgánicos. Estos posibles metalofármacos exhiben una alta versatilidad química en términos de farmacocinética y farmacodinámica.
R es uno de los lenguajes de programación más utilizados en quimioinformática, biología estructural e inteligencia artificial, entre otros (17,18). Algunas bibliotecas en R facilitan el preprocesamiento y la visualización de datos, el desarrollo de redes neuronales o el uso de computación paralela en los procesadores; las que usamos en este trabajo se mencionan específicamente a continuación. Tidyverse es un conjunto de paquetes en R diseñados para facilitar y mejorar el flujo de trabajo en análisis y manipulación de datos (19). Tidyverse proporciona un conjunto coherente y consistente de herramientas para realizar tareas comunes en análisis de datos, como tibble, ggplot2, entre otros. DoParallel es otro paquete en R que se utiliza para facilitar la ejecución paralela de tareas en múltiples núcleos de procesadores (20). La ejecución paralela implica realizar múltiples tareas simultáneamente, distribuyendo la carga de trabajo entre los diferentes núcleos de procesadores de una computadora. Esto puede conducir a mejoras significativas en la velocidad de ejecución y la eficiencia del programa, especialmente cuando se trata de operaciones intensivas computacionalmente. El paquete caret (Classification And REgression Training) en R es una herramienta versátil y poderosa que ayuda eficientemente en la creación, evaluación y ajuste de modelos de aprendizaje automático (21). Su objetivo es simplificar y estandarizar el proceso de desarrollo de modelos, desde la preparación de datos hasta la evaluación y selección del modelo. El paquete VIM (Visualization and Imputation of Missing Values) en R está diseñado específicamente para abordar el problema de los valores faltantes en los conjuntos de datos (22). Los valores faltantes son una ocurrencia común en el análisis de datos y pueden ser problemáticos, potencialmente introduciendo sesgos o afectando la calidad del análisis y los resultados del modelo. El paquete neuralnet en R es una herramienta utilizada para construir y entrenar redes neuronales artificiales (ANNs). Las ANNs son un tipo de modelo de aprendizaje automático inspirado en la estructura y funcionamiento del cerebro humano. Estas redes son capaces de aprender a partir de datos y realizar tareas como clasificación, regresión y reconocimiento de patrones. En este trabajo, proponemos la predicción de logP utilizando redes neuronales en el lenguaje de programación R, basada en una base de datos original compuesta por moléculas con una masa molecular superior a 500 Daltons. Estas moléculas son de interés en la química bioinorgánica medicinal debido a su alta afinidad por el hierro (sideróforos). La predicción del logP para sideróforos nos permitirá filtrar rápidamente moléculas con el potencial de llevar a cabo la estrategia farmacológica del efecto Caballo de Troya, que involucra la internalización de metales abióticos en microorganismos patógenos resistentes a los antibióticos.
PARTE EXPERIMENTAL
En este trabajo desarrollamos una base de datos de sideróforos y mostramos cómo a través de la generación de sus descriptores moleculares podemos predecir eficientemente su coeficiente de partición (LogP). Primero, hemos creado nuestra base de datos de 232 sideróforos mediante una búsqueda en la literatura científica en repositorios y páginas web. Se extrajeron los códigos SMILES (23) de las estructuras o se generaron si en caso no estaban disponibles. Para esta tarea nos apoyamos de Avogadro y además desarrollamos la página web https://quimicaorganica.streamlit.app. Esta web es de acceso libre y fue desarrollada usando recursos de código abierto. Usando el código SMILES generamos las coordenadas tridimensionales y pre-optimizamos las estructuras usando el algoritmo de descenso de gradiente y el campo de fuerza MMFF94 con Open Babel. Después de generar las 232 estructuras tridimensionales en formato MOL2, utilizamos el programa RFL-Score (24) para extraer 250 descriptores químicos utilizando los programas de quimioinformática Padel (25) y RDKit (26), donde nuestro atributo a predecir es el logP, indicado como MolLogP. Los paquetes de R: Tidyverse, doParallel, caret, VIM, neuralnet, y modelr, fueron empleados para realizar el preprocesamiento de datos, la eliminación recursiva de características (RFE), la separación de los datos (80% para el entrenamiento y 20% para la prueba), el desarrollo del modelo y el cálculo de métricas de evaluación, como ya hemos reportado previamente (27). Este procedimiento se resume en la Figura 1.
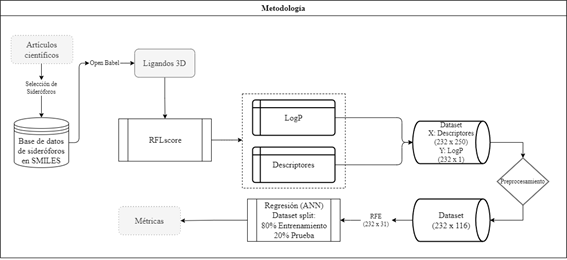
RESULTADOS Y DISCUSIÓN
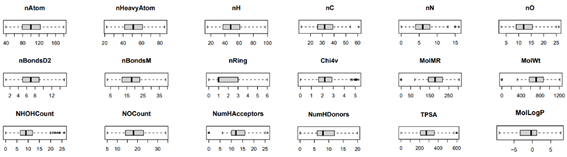
La base de datos moleculares en formato CSV (separado por comas), los descriptores generados, las estructuras tridimensionales, el modelo y los códigos implementados se encuentran disponibles en nuestro repositorio: https://github.com/inefable12/logP_ann. De los 250 descriptores obtenidos con Padel y RDKit (Figura 2), una gran cantidad estaba compuesta de ceros, por lo cual, desarrollamos un script para considerar las columnas con un mayor aporte de datos no nulos. De esta manera, conservamos únicamente 116 columnas de las 250 iniciales, que contenían al menos 150 datos diferentes de cero, lo cual significó una disminución del 46% del total de atributos. También se incorporó una columna con el tipo de sideróforo a la base de datos. Esta información es valiosa para posteriores investigaciones asociadas con el desarrollo de modelos de clasificación basado en descriptores moleculares.
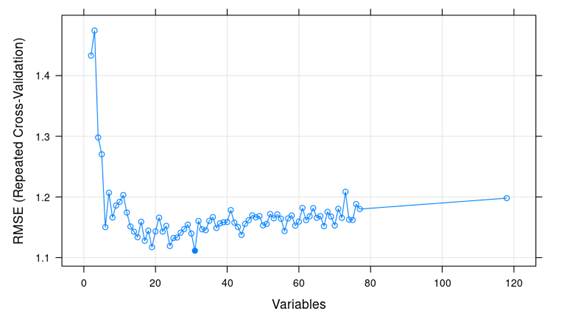
Durante la selección de atributos con RFE aplicada a los 116 atributos, se identificó un menor error cuadrático medio (RMSE) en la validación cruzada repetida cuando se utilizaron 31 atributos, como se destaca con un punto azul en la Figura 3. Estos corresponden a los descriptores: TPSA, PEOE_VSAn (n: 1, 6, 7, 8, 9, 10, 12), nN, SlogP_VSA5, SlogP_VSAm (m: 1, 6, 7), SlogP_VSA2, SA_EState7, MQNs_polarity_counts_hba, Chi3v, SMR_VSA5, MQNs_atom_counts_ao, NumHeteroatoms, Kappa3, NOCount, NumHDonors, nC, FractionCSP3, Chi2v, MQNs_atom_counts_c, SMR_VSA3, BalabanJ, SMR_VSA7, NHOHCount. En seguida los datos fueron normalizados y particionados en conjuntos de entrenamiento (80%) y prueba (20%) para la siguiente etapa.
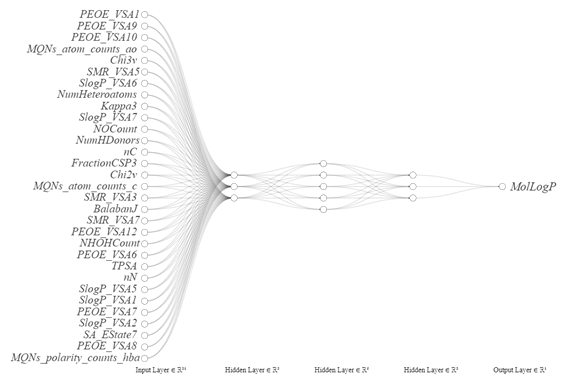
La arquitectura de la ANN consistió en 31 descriptores como entrada (input), 3 capas ocultas (3, 5 y 3 neuronas, respectivamente) y 1 salida, la predicción de LogP (Figura 4). Al representar gráficamente los datos de prueba, “LogP real” frente a los valores predichos o “LogP predicho” observamos la tendencia de estos valores (Figura 5). La regresión lineal nos da 0.99, 0.011, 0.00012, 0.0083 para las métricas R2, RMSE, MSE y MAE, respectivamente, lo cual es un indicador de la alta precisión del modelo, como se observa en la Figura 5. Empleamos un script para distribuir la ejecución de cálculos en los procesadores en paralelo, lo cual permitió poder explorar diferentes combinaciones de capas ocultas, número de neuronas y tasa de aprendizaje, sin depender del uso de supercomputación que es habitual en estos casos.
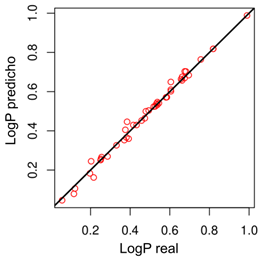
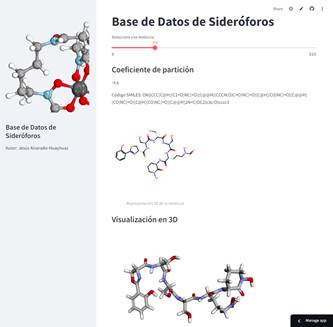
Si bien estamos demostrando la obtención de un modelo con excelentes métricas para la predicción del target, en general es requerido realizar una optimización de hiperparámetros lo cual requiere de computación de alto rendimiento, como se han reportado en otros trabajos. Tetko y colaboradores trabajaron la predicción de logP basado en el método de distribución atómica reportando un R2 de 0.918, utilizando un generador de descriptores comercial (MOE) y SMARTS, un codificador molecular más sofisticado que SMILES (18), mientras que otra publicación reciente basada en aprendizaje por transferencia predice el logP de pequeñas moléculas de un conjunto de datos de Reaxys y PhysProp con un R2 de 0.988 (13). Por lo tanto, destacamos nuestro procedimiento por tratarse de un modelo eficiente y constituido con herramientas de código abierto. Finalmente, la base de datos es también accesible desde https://sideroforos.streamlit.app, desde dispositivos móviles y computadoras en cualquier sistema operativo (Figura 6).
CONCLUSIONES
Este trabajo ha generado una base de datos única que incluye el código SMILES, las coordenadas atómicas tridimensionales y 250 descriptores moleculares de 232 sideróforos, lo que permitirá a otros investigadores trabajar más fácilmente en estudios de propiedades fisicoquímicas y farmacológicas de estas moléculas. Se ha demostrado que es posible predecir el coeficiente de partición octanol-agua (logP) de los sideróforos mediante redes neuronales artificiales con un alto grado de precisión (R² = 0.99). Este método es particularmente relevante, dado que el cálculo experimental del logP es costoso y la predicción computacional tradicional se ve limitada por la masa molecular y el tipo de átomos en las moléculas. A diferencia de otros estudios que dependen de herramientas computacionales costosas y bases de datos comerciales, este trabajo demuestra que es posible obtener modelos predictivos eficientes utilizando recursos computacionales domésticos y software de código abierto, lo que facilita su aplicación en una variedad de entornos de investigación. La base de datos y el modelo desarrollado proporcionan una herramienta valiosa para estudios de fármacos basados en el efecto Caballo de Troya, la simulación molecular y el desarrollo de nuevos modelos predictivos en química medicinal. Aunque se ha logrado una excelente precisión en la predicción del logP, destacamos también la necesidad de optimizar los hiperparámetros del modelo para obtener resultados aún más robustos, particularmente si se emplean técnicas de computación de alto rendimiento. Este estudio sienta las bases para investigaciones futuras en el campo de la quimioinformática y el diseño de fármacos basados en sideróforos, utilizando inteligencia artificial para abordar desafíos clave en la predicción de propiedades moleculares.

