











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCTION
Variegate porphyria (VP) is a rare metabolic disorder caused by an alteration in the porphyrin biosynthesis pathway, including the heme group of hemoglobin protein1.
This enzymatic alteration produces the deposition of heme precursors (porphyrin, 5-aminolevulinic acid [ALA] and porphobilinogen) in different tissues, mainly in the liver and erythrocytes2. Consequently, it can cause various non-specific symptoms such as abdominal pain, nausea, and vomiting. This clinical presentation and the low prevalence of the disease, makes diagnosis and proper management difficult3. VP generates acute neuro-visceral manifestations (abdominal, psychiatric, neurological or severe cardiovascular symptoms) with the risk of hepatic and renal complications4, and often triggers skin lesions in sun-exposed areas such as skin fragility, erosions, blisters, milia and pigmentary changes5.
In many cases the diagnosis is delayed for several years which may result in inadequate treatments and thus probably increase morbidity and the use of unnecessary medical resources. The following is a case of VP in a young Peruvian woman with a nonspecific clinical presentation and severe complications, which were associated with a mutation of the porphyrinogen oxidase enzyme gene (PPOX) not previously reported with VP.
CASE REPORT
A 21-year-old woman from the southern region of Peru, with ancestry from the Peruvian highlands, presented to a lower complexity hospital (Regional Hospital of Moquegua) complaining of 7 days of diffuse cramping abdominal pain of moderate intensity. The patient also presented emesis, constipation and asthenia. She reported a history of dysmenorrhea and consumption of nonsteroidal anti-inflammatory drugs (NSAIDs). She did not report any other personal or family history of interest. After ruling out a surgical cause for abdominal pain, the patient was admitted in hospitalization for further studies. During hospitalization, on the third day she presented moderate hyponatremia (129 mEq/L) and on the seventh day a generalized tonic-clonic seizure that evolved to status epilepticus, requiring management by an intensive care unit (ICU) (Figure 1).
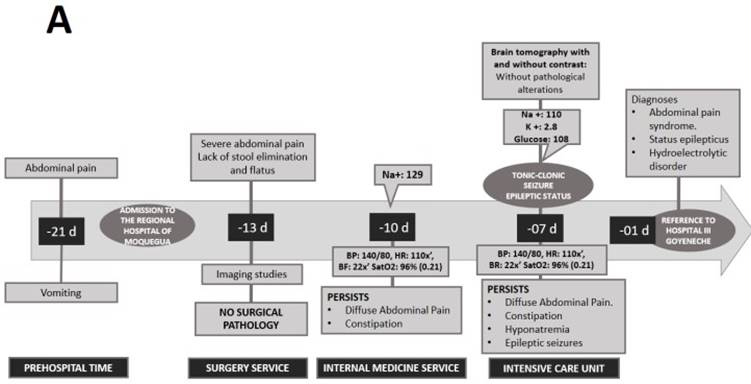
After stabilization, she was referred to the ICU of a higher complexity hospital (Hospital III Goyeneche) due to persistent abdominal pain, constipation for 8 days, red urine for 7 days and hydroelectrolyte disorder (hypokalemia and severe hyponatremia). Physical examination revealed poor hydration status and normal vital signs. The abdomen was painful on palpation with decreased sounds and no signs of peritoneal irritation. Neurological examination showed somnolence and ascending paresis of the lower extremities. The rest of the physical examination was normal, with no skin lesions or signs. Laboratory tests showed the following values: hemoglobin 14.2mg/dL, leukocytes 5 170 103/mm3, amylase 153 (U/L), lipase 204 (U/L), sodium 128 mEq/L, osmolarity 261 mOsm and lead 1.21ug/dL. Urinalysis showed red color, with 2-4 erythrocytes per field. Abdominal radiography showed mild dilatation of loops with hydroaerial levels, abdominal ultrasound showed signs of mild fatty liver and moderate intestinal ileus. The simple chest X-ray, abdominal and cerebral tomography did not show any alterations. With all these findings, the possibility of surgical, neoplastic, infectious pathology, acute pancreatitis or lead intoxication were ruled out (Figure 2).
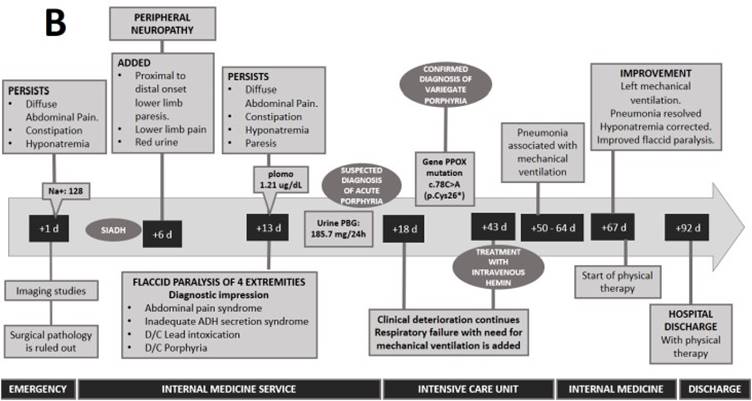
The alteration of sodium and osmolarity, together with the tonic-clonic coma, led to the diagnosis of inappropriate antidiuretic hormone secretion (SIADH). Likewise, the presence of red urine, paresis of lower limbs progressing to flaccid paralysis with respiratory failure led to the suspicion of acute porphyria. Therefore, urinary porphobilinogen (185.7 mg/24h) was requested, confirming the diagnosis of porphyria. To specify the type of porphyria, a genetic panel analysis for acute hepatic porphyrias was performed. Regarding this genetic panel, it includes deletion/duplication analysis and sequence analysis of the entire coding region for the following genes: ALAD, CPOX, HMBS and PPOX. In this case, it was reported a mutation in the enzyme porphyrinogen oxidase (PPOX variant; nucleotide change: c.78C > A and protein change: p.Cys26*) which confirmed the diagnosis of variegate porphyria (VP). Family screening by genetic testing or urine PBG analysis was not performed.
After administration of hemin arginate (25 days after diagnosis), the patient was weaned from mechanical ventilation and hyponatremia was corrected (Figure 2). However, recovery from paresis was slow and progressive, and after six months of rehabilitation the patient had limitations in lifting in her daily activities. After two years, the patient has no functional limitation in her daily activities and has not presented any disease flare-ups.
DISCUSSION
We present the case of a young patient with a confirmed diagnosis of VP after presenting an acute neuro-visceral crisis of porphyria, without cutaneous manifestations, with neurological and respiratory complications. The rare pathogenic variant of PPOX gen (PPOX variant; nucleotide change: c.78C > A and protein change: p.Cys26*) was identified.
In Europe, the prevalence of VP varies between 2.4 to 4 cases per million3 and since its initial description in 193718, more than 140 mutations have been identified. In South Africa, a higher prevalence has been reported, with 1 case per 300 births6,7. In contrast, in Latin America it is rare8,9. In Chile, 105 cases of acute porphyria have been reported over a 17-year period, with VP being the most frequently reported (21%)9. According to the available literature, we present the first case of VP reported in Peru.
VP presents an autosomal dominant inheritance, with mutation in the enzyme protoporphyrinogen oxidase (PPOX) which is the penultimate enzyme of the heme pathway10. VP affects more females and typically presents in adolescence or young adulthood4,9, as in our patient.
Regarding the clinical picture, although the cutaneous presentation (60-82%) is the most predominant3,4,10, its absence has been reported in 13% of cases of VP3 and has been associated with mutation of the I12T allele of the PPOX gene11. These photosensitivity symptoms are produced by the precipitation of heme precursors after exposure to UVA light12. Our patient came from Moquegua, a region in the southern coast of Peru with high exposure to UV rays13, therefore, the absence of cutaneous symptoms in VP was of great interest.
Regarding the acute neuro-visceral manifestations, it is known that they are typically nonspecific, the most common being the presence of acute, intermittent, diffuse abdominal pain, without peritoneal signs, which may or may not be accompanied by vomiting and constipation5. This coincides with the presentation of our patient. The presence of these acute attacks has been related to triggering factors (drugs such as barbiturates, contraceptives, alcohol and tobacco consumption, metabolic stress, among others)14. However, in our case there was no evidence of any of these factors.
The patient presented red urine, which was due to increased metabolic wastes related to PPOX enzyme deficiency1. The disorientation and tonic-clonic seizures could have been caused by the direct effect of the accumulation of porphyrins in the neurons or by the hyponatremia resulting from the SIADH consequent to VP15. Similarly, peripheral motor neuropathy in acute porphyrias generates neuromuscular weakness, from proximal to distal direction, with an increased risk of quadriparesis and respiratory failure15.
The diagnosis of VP is determined by increased urinary porphobilinogen (PBG) excretion and genetic confirmation12. In our patient, an elevated urinary PBG (185.7mg/24h) was found and the diagnosis of VP was confirmed by genetic analysis, which revealed a rare pathogenic variant (PPOX variant; nucleotide change: c.78C > A and protein change: p.Cys26*). This pathogenic variant has been previously identified in the literature in some cases of VP in Europe, but has not been reported in Latin American countries16,17,18.
The goal of treatment is to reduce hepatic ALA synthetase-1 (ALAS-1) activity, which can be regulated by carbohydrate administration (300 g glucose per day)2. However, the most effective therapy is the administration of intravenous heme, which results in the decrease of ALA and PBG in urine and plasma2,19, and in a clinical improvement in 4 days generally14. In the case presented, recovery of respiratory failure and hyponatremia was observed after hemin administration. In addition to a progressive recovery of muscle strength.
This case shows a particular presentation of VP and emphasizes the importance of considering the diagnosis even among patients with no family history, who do not present cutaneous manifestations and in countries where the incidence is practically unrecorded. Early diagnosis and treatment are of great importance to avoid life-threatening complications.
CONCLUSION
In conclusion, we report the case of a 21-year-old female patient with VP with acute symptoms of a severe neuro-visceral crisis without cutaneous manifestations and biochemical abnormalities, which was associated with a rare pathogenic variant (PPOX variant; nucleotide change: c.78C>A and protein change: p.Cys26*) in the PPOX gene. The importance of clinical investigations in patients with genetic mutations is essential to establish a genotype-phenotype correlation that facilitates the clinical approach and management in these patients. In addition to recognizing that timely diagnosis and treatment is essential to avoid seizures and severe complications.

