











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
INTRODUCCIÓN
El complejo esclerosis tuberosa (CET) es un síndrome multisistémico autosómico dominante de penetración variable causada por mutaciones en los genes supresores de tumores TCS1 en el cromosoma 9q34 o TCS2 en el cromosoma 16p13.3, los cuales codifican las proteínas hamartina y tuberina, respectivamente. Estas mutaciones provocan una disrupción en el regulador de proliferación celular diana de rapamicina en mamíferos (mTOR, mammalian target of rapamycin) favoreciendo el desarrollo de hamartomas en órganos como el corazón, cerebro, piel, riñones u ojos1.
Los tumores cardíacos primarios son extremadamente raros en los niños2. Se ha encontrado una incidencia del 0,027% al 0,08% en autopsias pediátricas2, mientras que en la vida fetal algunos estudios estian una incidencia de tumores cardíacos entre 0,05 a 0,14%3). Alrededor del 90% de estos tumores son benignos2. Desde el punto de vista histológico, los tumores benignos más frecuentes en etapa pediátrica son los rabdomiomas (60%), compuestos por las características células ‘en araña’, seguidos de los fibromas (12%), los mixomas (10%), los teratomas intracardiacos (25%) y los hemangiomas4.
En el presente estudio se comunica un caso de diagnóstico prenatal genético de esclerosis tuberosa en el contexto de un feto con múltiples rabdomiomas.
CASO CLÍNICO
Paciente de 39 años gesta 3, para 2 sin antecedentes médicos o familiares de importancia. Fue evaluada inicialmente en nivel de atención primaria. A las 24 semanas 4 días acudió a una clínica privada para ecografía morfológica de rutina donde encontraron 3 rabdomiomas a nivel intraventricular izquierdo, siendo la de mayor tamaño de 9 x 4 mm (a descartar esclerosis tuberosa) y refirieron a la paciente al servicio de medicina fetal de nuestra institución.
La paciente fue evaluada en el Instituto a las 28 semanas, cuando se confirmó las lesiones y se evidenció aumento en el número y tamaño: 3 en cavidad ventricular izquierda, (siendo la de mayor tamaño 11 x 7 mm), 1 en el septo interventricular y 2 en cavidad auricular derecha (6 x 7 mm y 6 x 9 mm, respectivamente) (figura 1). El bienestar fetal se encontró conservado con perfil biofísico 8/8 y Doppler fetal normal e índice de rendimiento miocárdico (IRM) 0,44, normal.
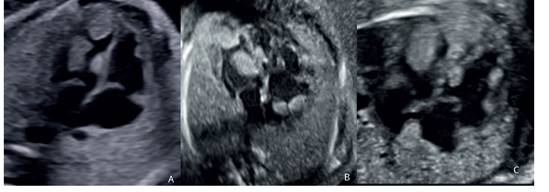
Figura 1 (a) Ecografía a las 24 semanas: radbomiomas en el ventrículo izquierdo. (b) 28 semanas: rabdomiomas en ventrículo izquierdo y aurícula derecha. (c) 31 semanas: rabdomiomas en ambos ventrículos y aurícula derecha. se logra visualizar aumento en el número de lesiones y en el tamaño con la edad gestacional.
Se explicó a la paciente la probable asociación sindrómica y previa consejería y consentimiento se procedió a amniocentesis a las 29 semanas para análisis en líquido amniótico de panel molecular de complejo esclerosis tuberosa y cariotipo.
A las 31 semanas se recibió los resultados de cariotipo 46,XX normal y se realizó ecocardiografía control con aumento en el número de lesiones y tamaño, extendiéndose hacia el ventrículo derecho. Así mismo, se notó aumento del índice de rendimiento miocárdico hasta 0.57, sobre el límite superior normal. No se evidenció obstrucción de los tractos de salida o entrada, regurgitación valvular ni alteración de las pruebas de bienestar.
En el control a las 33 semanas, los rabdomiomas incrementaron de tamaño generando disminución del llenado ventricular a predominio de lado izquierdo, con leve regurgitación mitral. Con relación al índice de rendimiento miocárdico, se encontró en 0,6, progresión a disfunción cardiaca no severa.
A las 35 semanas se recibió el resultado del panel molecular en líquido amniótico, que informó feto con heterocigosis de la variante patogénica NM_000548.4:C 1599+1G>A en el gen TSC2, confirmando una esclerosis tuberosa. La paciente fue hospitalizada por tener contracciones y se le realizó cesárea de emergencia con un recién nacido femenino de 2,595 g, Apgar 8 al minuto y 5 minutos, talla 44,5 cm.
Al examen físico del neonato, se halló máculas hipopigmentadas en piel (figura 2) compatibles con CET; la ecocardiografía confirmó los hallazgos prenatales. Fue evaluada por neurología y oftalmología sin encontrar hamartomas. Fue dada de alta al décimo tercer día de vida.
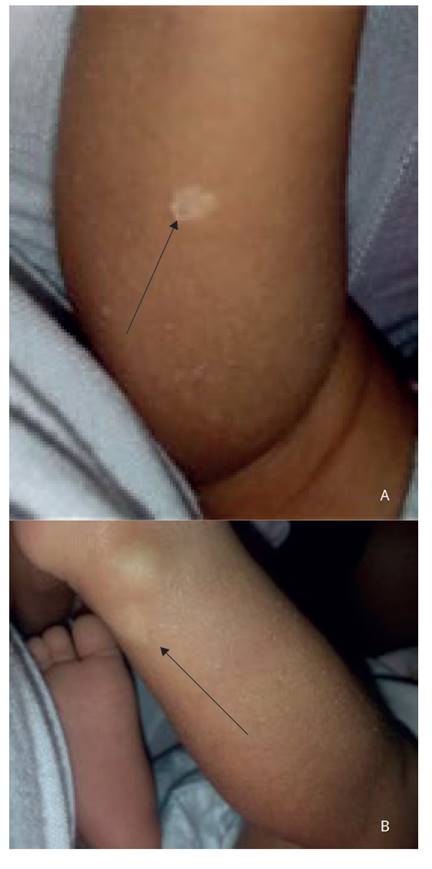
Figura 2 las flechas señalan máculas hipopigmentadas en a. pantorrilla y b. cara posterior del tobillo.
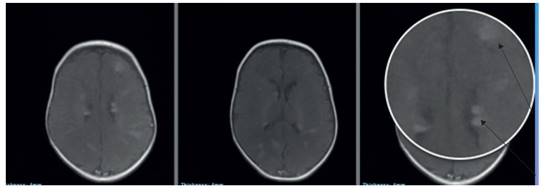
Al control del mes, la resonancia magnética cerebral mostró múltiples túberes corticales y subependimarios (figura 3). A los 6 meses de vida, la lactante permanece clínicamente asintomática y con disminución en el tamaño de los rabdomiomas a la ecocardiografía de control. En la evaluación familiar, el padre de la lactante presenta lesiones cutáneas de iguales características y actualmente está en estudio para confirmar CET.
DISCUSIÓN
La evidencia sugiere que los rabdomiomas son en realidad hamartomas de miocardio compuestos por células que se parecen a los miocitos cardíacos en lugar de verdaderas neoplasias5. Generalmente se presentan como tumoraciones pequeñas y múltiples, localizadas en su mayoría en el miocardio ventricular, aunque también se han descrito casos de afección auricular6. El sitio, el número y la obstrucción del tracto de salida ventricular de los rabdomiomas cardíacos se pueden observar en la vista apical de cuatro cámaras7.
El rabdomioma cardíaco fetal suele aumentar de tamaño hasta el nacimiento, luego de lo cual disminuye gradualmente y remite de manera espontánea en aproximadamente la mitad de los casos6,8, coincidiendo con la evolución del caso presentado. Aunque la expresión clínica es amplia, la mayoría son asintomáticos9, por lo que las manifestaciones clínicas dependerán de su tamaño y localización intracardiaca, pudiendo ocasionar desenlaces adversos como arritmias6, hidropesía fetal, muerte intrauterina y muerte súbita del lactante 8,10.
Los rabdomiomas cardíacos están presentes en el 47% a 67% de las personas con esclerosis tuberosa11, Por ello es fundamental que en la vida fetal, una vez identificado un rabdomioma cardiaco, se realicen estudios complementarios, en especial de diagnóstico molecular en búsqueda de esclerosis tuberosa.
La población pediátrica con esclerosis tuberosa tiene una probabilidad de hasta el 90% de tener manifestaciones neurológicas o neuropsiquiátricas, como epilepsia, déficit cognitivo y/o autismo12. La resonancia magnética fetal en el tercer trimestre de la gestación puede ayudar en el diagnóstico prenatal de esclerosis tuberosa al permitir la visualización de nódulos subependimales o túberes corticales que producen estas manifestaciones postnatales13,14). Sin embargo, la ausencia de lesiones cerebrales en una resonancia fetal no descarta esclerosis tuberosa15.
El confirmar el diagnóstico de esclerosis tuberosa con genética molecular en la etapa prenatal impacta en el pronóstico del paciente y su familia. En primer lugar, otorga herramientas para una consejería más precisa en el embarazo, en vista que el pronóstico posnatal de un rabdomioma aislado es diferente al pronóstico de fetos con rabdomiomas en el contexto de CET, por su predisposición a tener y desarrollar más hamartomas posnatalmente y el impacto neurológico.
Además, el identificar la mutación específica responsable de tuberosis esclerosa en el feto facilita la información sobre predisposición a manifestaciones específicas del CET. Por ejemplo, la mutación de novo en TSC2 es un factor de pronóstico negativo en comparación con mutación en TSC1(16). De igual manera, los pacientes con mutaciones grandes que afectan los genes de poliquistosis renal PKD1 tienen mayor predisposición a desarrollar astrocitomas subependimarios de células gigantes (SEGAs) a más temprana edad que otras mutaciones en el TSC21.
Así mismo, al ser la CET una condición de expresión clínica variable, el confirmar genéticamente el diagnóstico de esclerosis tuberosa en un feto con rabdomiomas permite extender la búsqueda de la enfermedad a los padres y hermanos, que pueden tener CET con manifestaciones clínicas no diagnósticas. Y permite que los padres tomen decisiones informadas de su futuro reproductivo, en caso tengan también la mutación en TCS1 o TCS2.
Adicionalmente, se ha informado que el diagnóstico prenatal del CET mejora el pronóstico neurológico al disminuir la ocurrencia de epilepsia en comparación a los diagnosticados posnatalmente, incluyendo mejoría significativa en áreas cognitivas, del lenguaje y motoras12, en especial en la población con esclerosis tuberosa en el espectro autista. Al prevenir la epilepsia o al mejorar la terapia para las convulsiones en la infancia se ayudaría a mitigar los síntomas de autismo en esta población17.
Se encuentran en estudio los inhibidores mTOR para la prevención y tratamiento de epilepsia en niños con CET18, lo cual aumenta la importancia en hacer el diagnóstico temprano de tuberosis esclerosa, en especial en la población asintomática.
En vista de la importancia del diagnóstico prenatal, en el 2022 Xiao-Yan Yang y colaboradores publicaron un estudio piloto sobre el uso de ADN fetal en sangre materna para el tamizaje de CET, con resultados similares a los obtenidos en pruebas invasivas o confirmatorias posnatales. Sin embargo, aún se requieren más estudios al respecto19.
En conclusión, se debe sospechar de esclerosis tuberosa fetal o familiar ante el hallazgo fetal de rabdomiomas cardiacos, aun si no hay manifestaciones clínicas evidentes en la familia. El pronóstico posnatal dependerá de la localización de los hamartomas y el grado de compromiso hemodinámico y se verá directamente impactado por el diagnóstico prenatal molecular al permitir mejorar el diagnóstico, vigilancia y manejo oportuno de este síndrome genético.

