











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La leucemia/linfoma T del adulto (LLTA) es una neoplasia linfoproliferativa T agresiva, producida por la infección del virus linfotropo T humano tipo 1 (HTLV-1). Es una enfermedad de mal pronóstico debido a quimio resistencia e inmunosupresión severa. Fue descrita por primera vez por Uchiyama et al. en una isla Japonesa1, y recién en los años 80 se evidenció un área endémica de HTLV-1 en el sudoeste de Japón. Pronto se identificaron otras áreas en el Caribe, Centro y Sud América, y Medio Oriente2. Se cree que existe alrededor de 1 millón de portadores de HTLV-1 en Japón y se estima 5 a 10 millones en el mundo2. En Japón, el riesgo estimado de desarrollar una LLTA en portadores del virus es 6% a 7% en varones y 2% a 3% en mujeres, y el tiempo de latencia para el desarrollo de ésta es 40-50 años, a diferencia de lo que ocurre con la paraparesia espástica y otras enfermedades no neoplásicas asociadas al HTLV-1, en las que el periodo para su desarrollo tras la infección es menor. La edad de comienzo de la enfermedad depende del área geográfica, así por ejemplo en pacientes de América del sur, Caribe y en Estados Unidos la edad de media de diagnóstico es de 40-50 años, mientras que en Japón es de 50 a 66 años3-6.
Por otro lado, en Japón hay un claro predominio en varones, mientras que en otras áreas como Jamaica o algunas zonas de EEUU existe un leve predominio de mujeres7,8.
EL VIRUS
Fue en 1979 cuando se aisló por primera vez el HTLV-1, desde muestras de linfomas cutáneos. Fue el primer retrovirus que se asoció a una neoplasia, sin embargo el descubrimiento del virus del VIH en la década de los 80´opacaría por años su estudio9,10. La transmisión del HTLV-1 se produce principalmente por 3 vías: madre-hijo (ya sea parto o amamantar), parenteral o sexual11-15. De ellas, la más importante ocurre a muy temprana edad: amamantar con leche materna16. Esta transmisión resulta en la portación del virus. La mayoría de los portadores serán asintomáticos el resto de su vida, y se estima que sólo un 5% desarrollará una LLTA17. Como ya se mencionó, esto ocurre generalmente en adultos, 20 a 30 años post infección18. La patogenia no es enteramente comprendida. Se cree que es un proceso que abarca múltiples factores, tales como factores genéticos, epigenéticos, o virales.12-19 El HTLV-1 requiere procesos transcripcionales complejos20. Tiene la habilidad de juntarse y fusionarse con las células diana (Linfocito T). Esto resulta en una fusión de su ADN con el de su huésped21. Luego ocurre la diseminación, mediante expansión clonal22. Es decir, la infección aguda produce la transmisión genómica de célula a célula y luego, en una fase crónica, ocurre la expansión clonal23-24. Como se mencionó, no se conoce exactamente la causa de la leucemogénesis. La LLTA resulta de la proliferación clonal de células infectadas. Las proteínas regulatorias Tax, Rex y HTLV-1 Basic Zipper Protein (HBZ) juegan un importante rol oncogénico, lo que lleva a la persistencia viral, estimulación, crecimiento y posterior desarrollo tumoral. Es decir, las células en latencia adquieren anormalidades genéticas, lo que finalmente lleva a LLTA25. Cabe destacar que además el virus HTLV-1 causa desregulación del sistema inmune del huésped, con aumento de activación de linfocitos T, haciendo a los portadores más proclives a infecciones26.
PREVENCIÓN DE LA INFECCIÓN
Teniendo en cuenta que la forma más frecuente de infección del virus es la administración de leche materna, su suspensión se ha convertido en la principal forma de prevenir la infección. En un estudio de 1987 en Nagasaki, en que se evaluó la infección del virus debido a lactancia materna, se comprobó que esta disminuyó de 26% a 2.7% al no permitir alimentar con leche materna a mujeres portadoras27. Desde entonces la recomendación para prevenir la infección por HTLV-1 es no alimentar con leche materna, o hacerlo solo durante 3 meses28. Esta medida, sin embargo, debe tomarse con cautela en países de bajos recursos, ya que podría aumentar la mortalidad infantil29. Se ha considerado varios otros aspectos para prevenir la infección por HTLV-1, las que han reducido la prevalencia del virus en áreas endémicas de Japón (Tabla 1)30.
Tabla 1. Formas de prevenir la infección del virus.
| a. Realizar tamizaje del HTLV-1 a donantes de sangre en los bancos de sangre y evitar la transmisión por transfusiones. |
| b. Solicitar a las madres seropositivas, no amamantar o hacerlo menos de 3 meses, ya que la lactancia prolongada aumenta el riesgo de transmisión vertical, debido al traspaso de linfocitos infectados al niño |
| c. Conducta sexual segura en los portadores; uso de preservativos, para evitar el contagio sexual. |
| d. Realizar serología HTLV‐1 a la madre, hermanos e hijos del paciente portador, y realizar consejería, si es que ésta resulta positiva. |
Hasta el momento, no hay una modo efectivo para prevenir al desarrollo de LLTA entre portadores del virus.
LEUCEMIA/LINFOMA T DEL ADULTO EN AMÉRICA LATINA
La gran diferencia de etnias en América Latina explican la gran diversidad de áreas en las que el HTLV-1 es endémico, incluso encontrándose diferencia entre regiones de un mismo país31. Existen 2 teorías sobre la llegada del HTLV-1 a América Latina: desde esclavos africanos, o hace millones de años por el paso del estrecho de Behring2. Para tener un punto de comparación, en Japón la LLTA corresponde al 25% de los linfomas T periféricos, en norteamérica al 2%, y en Europa al 1%30. América Latina y el Caribe constituyen una de las regiones del mundo con mayor prevalencia de infección por HTLV-1. Recientemente, Oliveira et al.32realizó una revisión sistemática de todas las publicaciones de LLTA en Centro y Sudamérica. Las regiones más afectadas incluyen Colombia, Perú, la región de Jujuy en Argentina y Chile, así como en Bahía, Brasil33. Se estima que la LLTA representa un 1,1% los linfomas no Hodgkin, llegando a un 5,5% en Perú, o 0,5% en Chile34. En México, sin mebargo, existe muy poca población infectada35. Los casos en que mayor prevalencia se ha observado es en Brasil, donde un estudio de 1995 de Pombo de Oliveira et al36,37, mostró que el 32,4% de los casos de síndromes linfoproliferativos T maduros diseminados estaba representado por LLTA en Rio de Janeiro. En Bahía, el 26,4% de los linfomas cutáneos estudiados, correspondió a LLTA. En Perú, Gotuzzo et al35y Beltran et al38, han publicado casos de LLTA. Alrededor del 10% de los casos de linfoma no Hodgkin del Instituto Nacional de Cáncer en Lima, están asociados a HTLV-1. En Argentina y Marín et al39, han descrito casos de LLTA, en especial en la provincia de Jujuy al Noroeste del país, donde la prevalencia de HTLV-1 es la más alta del país (3,5%). En Chile, un estudio publicado en 2003 por Cabrera et al.40describió 132 casos de síndromes linfoproliferativos leucemizados, en donde la LLTA fue la enfermedad más común (48%), sin embargo en 2012 un estudio de 195 casos consecutivos41de linfoma no Hodgkin mostró solo un caso de LLTA (0,5%).
CLASIFICACIÓN Y CARACTERÍSTICAS CLÍNICAS
La Organización Mundial de la Salud (OMS) en su última versión del 201642la clasifica como una neoplasia T madura. La clasificación de Shimoyama43distingue cuatro subtipos clínicos de LLTA (Tabla 1), con diferente presentación clínica y pronóstico: 1) indolente, 2) crónica, 3) aguda (o leucemia) y 4) linfoma. Los 2 primeros se consideran de curso indolente, mientras que el tipo aguda y linfoma son formas muy agresivas. Bittencourt et al.propuso además el tipo primario cutáneo tumoral, que generalmente es un subtipo de los indolentes. Este tendría clínica y pronóstico diferente44-46. Las principales manifestaciones clínicas de la LLTA se presentan en laTabla 2.
Tabla 2. Criterios diagnósticos de los 4 subtipos de LLTA según Shimoyama
| Caracteristicas | Indolente | Crónica | Linfoma | Leucemia |
| Frecuencia (%) | 5 | 5 | 25 | 65 |
| Recuento linfocitos (x109/l) | <4 | ≥4 | <4 | Elevado |
| Linfocitos T anormales (%) en SP | <5 | ≥5 | ≤1 | Elevado |
| LDH (UI/L) | <1,5 veces VN | <2,5 veces VN | Elevada | Elevada |
| Calcio (mg/dL) | normal | normal | Elevado | Elevado |
| Compromiso piel y/o pulmón | ± | ± | ± | ± |
| Adenopatías | No | ± | Si | ± |
| Hepatoesplenomegalia | No | ± | ± | ± |
| SNC/óseo/pleura | No | No | ± | ± |
SP: sangre periférica, VN: valor normal.
Manifestaciones clínicas
El curso clínico es muy heterogéneo. Los pacientes pueden referir fatiga, adenopatías, hepatoesplenomegalia, LDH elevada, hipercalcemia e infecciones oportunistas. Las lesiones cutáneas están casi siempre presentes, pueden ir desde eritrodermia generalizada, placas, pápulas o nódulos rojizos, son siempre múltiples y frecuentemente generalizadas. Otras manifestaciones clínicas de LLTA menos frecuentes incluyen: derrames pleurales y ascitis, compromiso del sistema nervioso central (SNC), ya sea de pares craneanos o meníngeo. Las infecciones bacterianas, fúngicas o por gérmenes oportunistas son frecuentes, debido a inmunodeficiencia, la que es exacerbada por la quimioterapia. De forma infrecuente puede comprometer el intestino.
Agresivos: Aguda y linfoma
El subtipo leucemia o agudo, se presenta con síntomas consuntivos, células neoplásicas circulantes, adenopatías, hepatoesplenomegalia, lesiones óseas, cutáneas, hipercalcemia e infecciones oportunistas47. El tipo linfoma se presenta con adenopatías, sin células circulantes. Los pacientes pueden presentar lesiones cutáneas, pulmonares, visceromegalia e hipercalcemia, pero en menor medida que el tipo agudo43.
Lamentablemente se consideran las formas más frecuentes, tanto en América Latina como Japón, representando 55-60% los casos tipo agudo y 20-25% los tipo linfoma37,43,48. Sin embargo se debe destacar que en el International Peripheral T-cell Lymphoma Project se mostró que un 87% de los tipos agresivos fueron tipo linfoma49, lo que habría que estudiar con mayor detención.
Indolente y crónico.
El tipo crónico puede presentar linfocitos patológicos circulantes, y ocasionalmente pueden presentar lesiones cutáneas, pulmonares, adenopatías o visceromegalia. No se asocia a hipercalcemia ni compromiso de sistema nervioso central, huesos o tracto gastrointestinal43,47. Puede a su vez diferenciarse en subtipos favorable o no favorable, según albúmina y niveles de LDH. La importancia de diferenciar estas dos variables radica en que la primera no requiere tratamiento y la segunda sí. El tipo indolente muestra característicamente lesiones cutáneas o pulmonares, sin otro compromiso. Puede tener células patologicas en sangre periférica, pero estas son menos del 5%43,47.
Diagnóstico
Clínicamente, el diagnostico de LLTA no es difícil, y se hace basado en la seropositividad para HTLV-1, sumado a una neoplasia T madura concordante, comprobada por histología y/o citología47.
Seropositividad de HTLV1
El método más comúnmente usado es el de ELISA. Sin embargo, éste no diferencia entre HTLV-1 y 2, ya que estos 2 virus comparte un 70% de sus secuencias genómicas. Para confirmar la infección, se usa Western blotting50.
Sangre periférica.
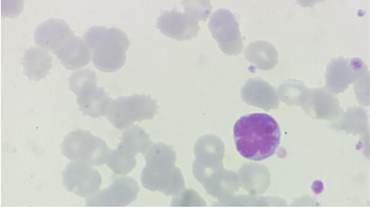
El diagnóstico se sospecha por la detección de los linfocitos de núcleo multilobulado en la sangre periférica. Estas células llamadas “tipo flor” o “flower cells” son linfocitos maduros que tienen el núcleo lobulado, cromatina gruesa, pequeño o sin nucléolo, y citoplasma agranular y basófilo51(Figura 1). Estas células se consideran patognomónicas de LLTA. Sin embargo, existe una gran diversidad morfológica. Es frecuente observar inmunocitos circulantes. En las formas indolente y crónica, los linfocitos son menos pleomórficos, más pequeños, de núcleo bi o multilobulado, de cromatina compacta. Más de 5% de linfocitos T anormales en sangre periférica por citología e inmunofenotipo bastan para hacer el diagnóstico de LLTA en pacientes sin lesiones tumorales.
Inmunofenotipo
En la mayoría de los pacientes, las células en la LLTA expresan un fenotipo de células T maduras, y son positivas para CD2, CD5, CD25 y CD45RO, T-cell receptor αβ y HLA-DR. Además son negativas para CD7 y el CD3 puede ser débil. Cerca del 90% de los casos son CD4 positivo y CD8 negativo42,52. El receptor 4 de quimokina (CCR4) es expresado en más del 90% de los casos y está asociado a mal pronóstico.
Médula ósea.
Generalmente no se requiere hacer un aspirado o biopsia de médula ósea para el diagnóstico. Sin embargo, puede dar información útil sobre sobre la reserva de médula ósea, antes de la quimioterapia.
Biopsia de sitio afectado.
Cuando el diagnóstico no puede realizarse en sangre periférica, se recomienda la biopsia ganglionar excisional, no por punción. Sin embargo, la histología de la piel y el ganglio no son específicas de LLTA.
El estudio de inmunohistoquímica demuestra que las células son positivas para CD3, CD5, CD25 y generalmente CD4, y negativas para CD7 y CD20. Aproximadamente un 5% son positivas para CD842.
Alteraciones bioquímicas
La hipercalcemia es la alteración de laboratorio más característica de la LLTA, a diferencia de otros síndromes linfoproliferativos crónicos. Se observa en la mitad de los casos de la forma aguda y en 20% de la forma linfoma. No se observa en las formas indolentes ni crónica. La LDH y β2 microglobulina séricas elevadas son alteraciones frecuentes y reflejan actividad/masa tumoral. En forma similar, está elevada la forma soluble de la cadena α del receptor de interleukina (IL)-2.
Imágenes radiológicas y endoscopía.
La tomografía axial computada de cuello, tórax, abdomen y pelvis es necesaria para detectar sitios de compromiso nodal o extranodal pe la enfermedad. Debe considerarse una endoscopia digestiva alta con biopsia, debido a que es frecuente el compromiso del tubo digestivo en la LLTA agresiva. La evaluación del SNC por imágenes y/o punción lumbar por compromiso de cerebro/meníngeo por LLTA o por infecciones oportunistas debe considerarse.
Estratificación riesgo
Se han reportado 3 sistemas de estratificación pronóstica en esta enfermedad.: ATL-PI, JCOG-PI y ATL-PI modificada. La primera fue validada en una cohorte japonesa de 807 pacientes con tipo linfoma y agudo. Esta dividió a los pacientes en 3 grupos, mediante 5 factores: estadío Ann Arbor, Performance Status (PS), edad, albumina, y receptor soluble IL-253. El score JCOG-PI analizó 276 pacientes enrolados en diferentes estudios del grupo japonés: Japan Clinical Oncology Group - Lymphoma Study Group (JCOG-LSG). Este dividea los pacentes en 2 grupos: Alto riesgo cualquiera que tuviera 1 ó más de los siguientes factores: PS>2, Calcemia elevada54. La ATL-PI modificada analizó 1792 pacientes. Las variables evaluadas fueron tipo agudo, PS pobre, altos niveles de recetor soluble de interleukina 2 (>5000U/mL), calcemia corregida >12 mg/dL, y niveles de proteína C reactiva > 2,5mg/dL.55 En América Latina no contamos con la medición de niveles de receptor soluble de IL-2 en muchos países, por lo que no serían una alternativa viable. Un score pronóstico más fácil de realizar en nuestra realidad es el IPI, que ya ha sido validado en otros estudios en pacientes con LLTA tipo linfoma49.
Pronóstico
En general, la LLTA se caracteriza por un mal pronóstico, con sobrevidas cortas56. Uno de los factores que más contribuyen es la quimioresistencia y la inmunosupresión asociada a LLTA, especialmente en sus formas agresivas57.
En Japón, la sobrevida global media bordea los 12 meses, incluso con quimioterapias intensivas58. Un estudio realizado por Katsuya et al59reportó 1594 pacientes tratados con terapias actuales. Este reportó una sobrevida media de 8.3, 10.6, 31.5, y 55 meses para la forma aguda, linfomatosa, crónica e indolente respectivamente. En otro estudio realizado en Nueva York se reportó una sobrevida media de solo 24 semanas5, y en un estudio retrospectivo la sobrevida a 5 años fue de 14%56.
Tratamiento
La mayoría de los pacientes no logran curarse con las opciones terapéuticas actuales, y la sobrevida no ha mejorado sustancialmente en los últimos 20 años. Debe tenerse en cuenta además que la mayoría de los estudios clínicos son realizados en población japonesa, por lo que no sabemos si son estudios completamente extrapolables a nuestros pacientes. De hecho, se han observados diferencias en resultados al comparar con población de estudios conducidos en EEUU60.
Opciones terapéuticas
Las opciones terapéuticas actuales para LLTA incluyen: observación hasta la progresión (watch and wait), interferón alfa (IFN), zidovudina (AZT), quimioterapia combinada, trasplante alogénico de progenitores hematopoyéticos y nuevos agentes. Sin embargo, en la actualidad no existe un tratamiento estándar para la LLTA. Tanto es así, que antes que cualquier recomendación específica, se incita a probar con estudios clínicos61.
Observación
Los pacientes con las formas indolente y crónica, pueden sobrevivir uno o más años sin QT. Se acepta que en formas indolentes podría usarse esta estrategia de observación activa51.
Quimioterapia convencional.
La quimioterapia más utilizada es la llamada CHOP-like, especialmente en la forma linfomatosa. Entre 1970 y 1980 se trataban igual que otros linfomas T, llegando a sobrevidas de 8 meses. En 1998 se realizó un estudio fase III62que comparó CHOP 14 (ciclofosfamida, doxorrubicina, vincristina, y prednisona), con VCAP-AMP-VECP (VCAP: vincristina, ciclofosfamida, doxorrubicina, y prednisolone; AMP: doxorrubicina, ranimustina, y prednisona; VECP: vindesina, etoposido, carboplatino, y prednisona). Si bien el segundo tuvo mayor toxicidad, logró mayor respuestas completes (40% vs 25%, P = 0.02) y mayor sobrevida global a 3 años (24% vs 13%). Por esta razón se considera estándar en la actualidad, sin embargo, muchas de las drogas de este esquema no se encuentras en América Latina. Dado el alto porcentaje de compromiso de SNC se recomienda profilaxis intratecal junto con la QT63.
Terapia antiviral e interferón alfa
Numerosos estudios han utilizado la combinación de un agente antiretroviral, zidovudina (AZT) e IFNα (AZT/IFN)64-67. Un reciente metaanálisis68mostró sobrevidas similares a las obtenidas con quimioterapia en formas no linfomatosas, incluída la forma aguda. En este se evaluaron 254 pacientes con LLTA tratados en USA, Reino Unido, Francia y Martinica, entre 1995-2008. El estudio demostró beneficio de una intervención temprana en LLTA tratados con IFNα/AZT. La sobrevida global (SG) a 5 años fue 46% para 75 pacientes que recibieron terapia antiviral en primera línea, 20% para 77 pacientes que recibieron QT en primera línea y 12% para 55 pacientes que recibieron QT en primera línea seguido de terapia antiviral. Los pacientes con LLTA crónica, indolente y aguda se beneficiaron significativamente de la terapia antiviral en primera línea, mientras que los pacientes con linfoma no se beneficiaron de esta estrategia. En los pacientes con LLTA indolente y crónica la SG a 5 años resultó de 100%, en la forma aguda fue 28% comparada con 10% de aquellos tratados con QT en primera línea. En cambio en pacientes con linfoma la SG a 5 años fue 0% comparada con 18% de aquellos tratados con QT en primera línea. Si bien tiene regular tolerancia, las guías occidentales recomiendan esta combinación en LLTA, excepto en el tipo linfoma. Sin embargo, las guías japonesas no lo recomiendan de rutina por escasa evidencia69.
Trasplante de progenitores hematopoyéticos.
El trasplante autólogo no ha mostrado tener utilidad en este tipo de pacientes, debido a una alta tasa de recidiva70. El trasplante alogénico, sin embargo, ha mostrado ser capaz de inducir sobrevidas a largo plazo en un 25-40% de los pacientes. Sin embargo la alta toxicidad de este procedimiento, de hasta 40% de mortalidad, hace tomar esta estrategia con cautela. En un estudio retrospectivo se mostró un análisis de 586 pacientes en que se realizó trasplante alogénico. De ellos, un 36% se mantenía vivo a los 3 años, con una SG media de 9,9 meses. Además, hubo una leve tendencia a una mejor sobrevida en pacientes mayores con régimen de intensidad reducida71. En otro estudio retrospectivo japonés se mostró una SG a 3 años de 33% en pacientes con trasplante alogénico72. Por otro lado, existen datos de una reciente revisión sistemática, que mostró 73% de remisión completa en estos pacientes, pero con altas tasas de recaída73. Por estos resultados, la American Society for Blood and Marrow Transplantation, en sus guías 2017, recomienda trasplante alogénico de primera línea en pacientes jóvenes con LLTA tipo aguda o linfoma74. En resumen, para LLTA crónica o indolente, puede ser apropiado la observación en pacientes asintomáticos. Si son sintomáticos podría tratarse con terapia local (dermatológico) o AZT/IFN. En tipo crónico se ha visto que la quimioterapia puede empeorar su curso, en comparación con observación75. Para tipo crónico no favorable y tipo aguda se recomienda enrolar en estudio clínico. Si esto no es posible, tratamiento con AZT/IFN o quimioterapia es opción.
En tipo linfoma se prefiere ingreso a estudio clínico. De no ser posible, se recomienda regímenes CHOP-like62.
Anticuerpos monoclonales
Recientemente fue aprobado en Japón el anticuerpo monoclonal (AcMo) anti-CCR4 (mogamulizumab) que actúa contra el CC cimokina receptor 4 (CCR4) que es expresado en las células neoplásicas de la mayoría de los pacientes con LLTA76-78. Su uso principalmente es en pacientes en recaída o refractario por este momento. Es este escenario mostró tasas de respuestas de 11% vs 0% en pacientes con otras terapias definidas por el médico tratante79.
CONCLUSIÓN
La LLTA es todavía una enfermedad de mal pronóstico, peor que otros linfomas T. No se observa plateau en la curva de sobrevida con los tratamientos actuales, en las formas agresivas o indolentes de LLTA, tratados con QT u observación y/o AZT/IFN. El uso de anticuerpos monoclonales como mogamulizumab asociado a QT intensiva ha demostrado mejoría en la sobrevida de pacientes con LLTA agresiva. El trasplante alogénico puede lograr cura en un reducido número de pacientes jóvenes, a pesar de una considerable mortalidad relacionada al trasplante. Se espera que nuevos agentes puedan incorporase al tratamiento de esta grave enfermedad. Mientras tanto, los esfuerzos para prevenir la diseminación del HTLV-1 deben maximizarse, realizando un gran esfuerzo de salud pública para mejorar la vigilancia del virus, con el tamizaje en los bancos de sangre y solicitarlo de rutina en el control del embarazo para reducir sustancialmente la trasmisión vertical del virus.

