











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La hipoacusia congénita tiene una prevalencia de 1 a 2 por cada 1000 niños al nacer y tiene consecuencias negativas para el lenguaje, el habla, las habilidades cognitivas y socioemocionales (1). La hipoacusia que presentan los pacientes con microtia y atresia aural son de tipo conductiva o mixta en la mayoría de los casos (2).
Según la Organización Mundial de la Salud, más del 5 % de la población mundial padece de una pérdida de audición discapacitante y requiere rehabilitación (34 millones de niños) (3).
La microtia es un defecto congénito del pabellón auricular al nacer, que va desde no estar desarrollado completamente hasta su ausencia completa, y puede ocurrir de forma aislada o asociada con otra malformación (2,4,5). Un paciente con microtia, sin síndrome asociado, puede vivir generalmente una vida normal y productiva (2).
La presentación más común de microtia es el grado III, que consiste en la hipoplasia de todo el marco cartilaginoso con un remanente lobulillar. (véase figura 1) (6).
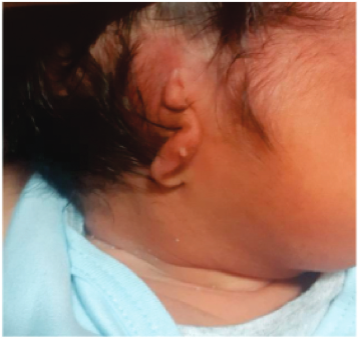
Figura 1: Paciente con microtia grado III de Meurman en oído derecho. Fuente: Instituto Nacional Materno Perinatal.
La atresia aural es un trastorno caracterizado por varios grados de hipoplasia congénita del conducto auditivo externo y está presente frecuentemente con microtia (6,7).
Se clasifican según los siguientes tipos:
Tipo A (estenosis): La porción fibrocartilaginosa y ósea del conducto auditivo externo están presentes, pero son estrechas.
Tipo B (atresia parcial): Solo algunas partes de la porción fibrocartilaginosa u ósea del conducto auditivo externo están presentes con la membrana timpánica rudimentaria.
Tipo C (atresia total): La porción fibrocartilaginosa y ósea del conducto auditivo externo, así como la membrana timpánica están ausentes (6).
El objetivo de este trabajo fue revisar y recopilar la evidencia actual en microtia y atresia aural congénita.
METODOLOGÍA DE BÚSQUEDA Y SELECCIÓN DE RESULTADOS
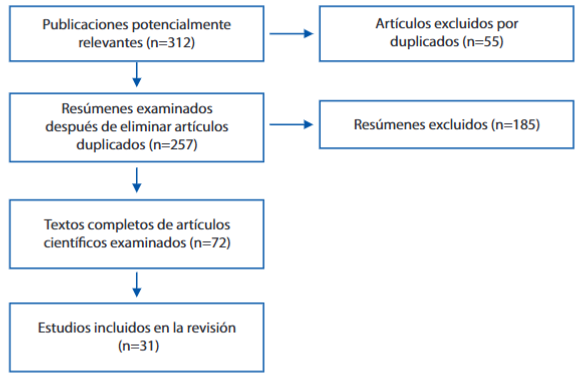
La revisión fue efectuada mediante búsqueda bibliográfica electrónica basado en evidencia con artículos en inglés y español en los sitios de búsqueda PubMed/Medline, Scopus, Clinical Key, SciELO, a partir del año 2010 hasta el año 2023 (véase figura 2) Se buscaron todas las referencias bibliográficas en español e inglés con las palabras clave: microtia congénita y atresia aural congénita. Se evaluaron artículos de revisión e investigación, que, en general, tenían menos de cinco años de publicados. Una vez identificados los artículos de interés, se consideraron como criterios de inclusión: 1. Que examinaran el diagnóstico y tratamiento de la microtia y atresia aural congénita, 2. que abordaran la temática de acuerdo con una metodología sistematizada (cuantitativa, cualitativa, otras). Luego de una evaluación del texto completo, se seleccionaron 31 estudios que se muestran en este artículo. Se tomó un periodo de cuatro meses (febrero 2023 a junio 2023) para la revisión. El presente trabajo se encuentra en las Líneas de Investigación de la Universidad Ricardo Palma 2021-2025 (31).
Desarrollo del tema
EPIDEMIOLOGÍA
Tiene una incidencia de 1 a 10 por 10 000 nacimientos (6-12), además de una gran connotación social, ya que las personas afectadas pueden sufrir de problemas psicológicos derivados de este defecto, visible y difícil de ocultar (13). La microtia está asociada a atresia aural en el 75 % de los casos (9). En México, se ha reportado una prevalencia de 7.37/10 000 nacidos vivos (14). En Perú, no existen datos nacionales que determinen la prevalencia de la microtia y atresia aural (15).
Puede ser unilateral o bilateral (29). El 90 % de los casos de microtia son unilaterales, el oído derecho es el más afectado que el oído izquierdo (5,6,11,12,16) y es más frecuente en hombres que en mujeres (5-7,12). Esta malformación está asociada con un síndrome en 30-60 % de pacientes (6, 17).
En los casos unilaterales, la microtia se define como una discrepancia de tamaño entre las orejas que excede la variación normal (18). La microtia bilateral se define como una longitud del oído externo que está más de dos desviaciones estándar por debajo de la media. En casos severos, el pabellón auricular está completamente ausente (anotia).
EMBRIOLOGÍA Y ANATOMÍA
El ectodermo somático interviene en la formación del oído externo e interno y constituyen los elementos epiteliales del pabellón auricular, conducto auditivo externo, la capa externa de la membrana timpánica y el laberinto membranoso del oído interno (19).
El pabellón auricular se desarrolla a partir de las tres prominencias auriculares del primer arco branquial (13,20). Es formada de la fusión de las seis yemas mesenquimatosas o prominencias auriculares de His en la superficie del embrión durante la quinta semana de desarrollo intrauterino. Su desarrollo termina hacia la décima segunda semana (21).
La anatomía del pabellón auricular se muestra en la figura 3.
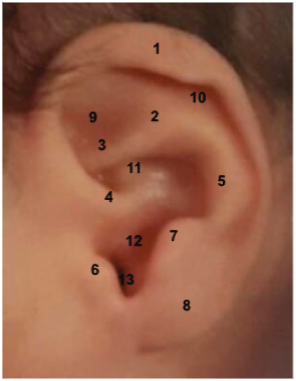
Figura 3: Anatomía del pabellón auricular. 1. Helix, 2. Cruz superior, 3. Cruz inferior, 4. Raíz del hélix, 5. Antihelix, 6. Trago, 7. Antitrago, 8. Lóbulo, 9. Fosa triangular, 10. Fosa escafoidea 11. Concha cymba 12. Concha cavum, 13. Muesca intertragal. Fuente: Instituto Nacional Materno Perinatal (INMP).
El conducto auditivo externo se forma a partir de la 8.a semana y su epitelio se desarrolla del primer surco branquial (22). Mide 2.5 cm, se extiende desde la concha hasta la membrana del tímpano y está formado por dos zonas:
Tercio externo cartilaginoso
Dos tercios internos óseos (22)
Tiene forma de S y se dirige, en primer lugar, hacia adentro, adelante y ligeramente hacia arriba; luego, discurre hacia atrás y arriba, y, por último, hacia adentro, adelante y algo abajo. 22
Las alteraciones precoces de este proceso originan una anotia o una microtia, mientras que las alteraciones más tardías, una malformación leve del pabellón auricular.
Las anomalías congénitas del oído externo y medio afectan estructuras derivadas principalmente del primer y segundo, por arcos branquiales, de la primera hendidura y de la primera bolsa faríngea.
ETIOPATOGENIA
Las causas no se encuentran completamente comprendidas y siguen siendo poco claras (17, 23). Factores genéticos y ambientales pueden generar una malformación durante la embriogénesis del oído externo, medio o interno principalmente durante la tercera y décima semana de gestación (19).
Embriológicamente, ocurre una falta de desarrollo y/o migración del componente mesodérmico y cartilaginoso (23).
La atresia del conducto auditivo externo se debería a la ausencia de reabsorción del tapón meatal o a un hiperdesarrollo del cartílago de Reichter (segundo arco branquial) (19). Las malformaciones del martillo y yunque pueden tener origen en una alteración de la diferenciación del cartílago de Meckel (primer arco branquial) que da lugar a una malformación de los huesecillos o a una fijación anómala del martillo y yunque (23).
FACTORES ASOCIADOS
El origen étnico, sexo masculino, bajo peso al nacer, enfermedad viral materna aguda, nivel educativo de la madre, diabetes materna, múltiples nacimientos, el consumo de talidomida, retinoides, aminoglucósidos, alcohol y tabaquismo durante el embarazo son factores asociados a microtia (9, 23, 24, 29). Asimismo, la infección por rubeola, citomegalovirus o toxoplasma gondii, el trastorno metabólico como el hipotiroidismo o cretinismo endémico, constituyen factores asociados a microtia (23, 24). Se ha encontrado que niveles más altos de ingesta de folato durante el embarazo reduce la incidencia de microtia (24). En menos del 50 % de pacientes con microtia se asocian a los siguientes síndromes: microsomía craneofacial, síndrome de Treacher Collins, síndrome de Goldenhar, síndrome de Crouzon, síndrome de Moebius, síndrome de Fanconi, síndrome de DiGeorge, síndrome de Pierre Robin, síndrome de CHARGE, síndrome de VACTERL, aplasia laberíntica, síndrome branquio-oto-renal (7, 9, 23, 24). Además, existe un estudio que asocia la microtia con la altitud (>2000 m. s. n. m.) en las ciudades (28).
CLASSIFICATIONS
Tanzer (27) y Meurman (25) clasificaron la microtia (véase tablas 1 y 2).
Tabla 1 Clasificación de Tanzer.
| I | Anotia (véase figura 3) |
| II | Hipoplasia completa (microtia): Con atresia de conducto auditivo externo Sin atresia de conducto auditivo externo |
| III | Hipoplasia de 1/3 medio de pabellón |
| IV | Hipoplasia de 1/3 superior Oreja en corneta o en taza (véase figura 4) Criptotia |
Tabla 2: Clasificación de Meurman.
| I | Pequeño pabellón, pero armonioso |
| II | Restos vestigiales del pabellón, conducto auditivo externo atrésico |
| III | Ausencia casi completa del pabellón, resto en forma de lóbulo |
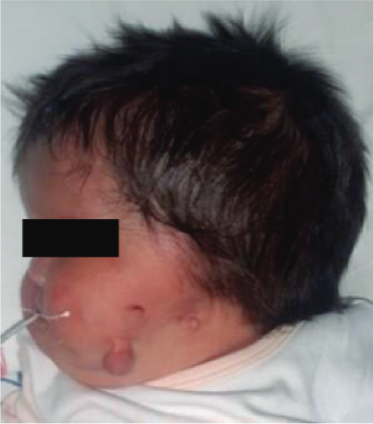
Figura 4. Paciente con anotia en oído izquierdo más melotia Fuente: Instituto Nacional Materno Perinatal.
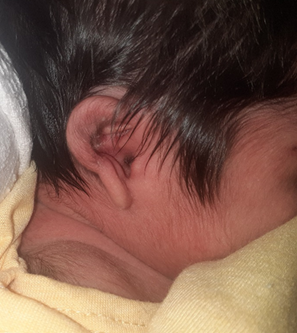
DIAGNÓSTICO
El diagnóstico de la microtia y atresia aural es clínico, apoyado de los exámenes complementarios. En el examen clínico, se debe observar la forma del pabellón auricular (orejas más pequeñas de lo normal), su implantación y estigmas (fístulas, apéndices o mamelones) (23, 29); examinar el meato, conducto auditivo externo (anormalmente estrecho, bloqueado o ausente) y el tímpano (23, 29); asimismo, es importante examinar la articulación temporomandibular (displasia de tejidos blandos) y rama ascendente del maxilar inferior, evaluar el aspecto y la conformación de las suturas del cráneo. Se debe evaluar, también, las asimetrías de la cara, hipoplasias del maxilar superior o inferior, apertura bucal, hendiduras palatinas o fisura submucosa y las características del cuello, tórax y miembros superiores e inferiores, presencia de quistes branquiales (23).
EXÁMENES COMPLEMENTARIOS
Las emisiones otoacústicas se indica para evaluar el oído sano. Un micrófono en el canal auditivo externo detecta estas emisiones otoacústicas de baja intensidad (1).
El pediatra o médico de familia tiene el primer contacto con el niño y debe conocer los factores de riesgo para hipoacusia y el tamizaje auditivo. El otorrinolaringólogo debe poseer todos los equipos y tener formación en diagnóstico infantil.
Los potenciales evocados auditivos son los exámenes complementarios de elección. Los potenciales evocados auditivos de tronco encefálico (PEATC) o BERA (1); permiten medir a nivel del tronco cerebral la respuesta eléctrica de la vía auditiva, que incluye la cóclea y la vía retrococlear, con electrodos de superficie. Asimismo, los potenciales evocados auditivos de estado estable (ASSR) miden el nivel de audición y permiten evaluar la vía ósea.
La audiometría a campo libre, por juego y tonal, están indicados según la edad del niño (21, 25).
La timpanometría está indicada en conductos permeables o en el oído contralateral a la disgenesia para determinar posibles malformaciones de la cadena osicular, en oídos aparentemente normales (23).
En cuanto a los estudios de imágenes, se solicita tomografía computada de peñascos (oídos) con cortes finos axiales y coronales sin contraste. Permite valorar el hueso temporal, evaluar el hueso timpanal y la mastoides, la caja del oído medio, su relación con el nervio facial, la cadena osicular y la conformación del laberinto óseo.
La tomografía computada de peñascos se realiza alrededor de los cinco a seis años, o antes en casos de disgenesia bilateral o de sospecha de colesteatoma (25). La resonancia magnética, específicamente de la fosa posterior, se solicita para evaluar la conformación de las estructuras membranosas del caracol, laberinto posterior y pares craneales.
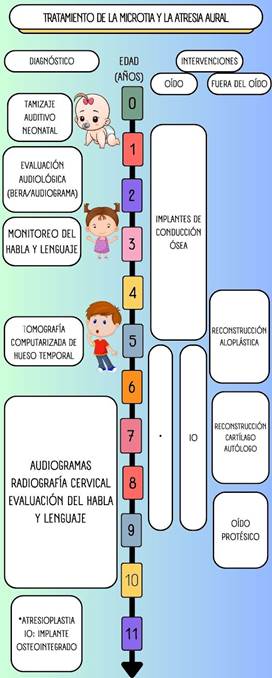
TRATAMIENTO
Se indica un dispositivo vibrador óseo tipo vincha o banda suave antes de que los pacientes puedan ser candidatos para la cirugía de vibrador óseo osteointegrado para estimular el nervio auditivo (34). El uso de los dispositivos de conducción ósea en pacientes jóvenes ayuda en la adquisición de las habilidades lingüísticas en los periodos críticos de la vida (34).
Las opciones reconstructivas para la microtia son los siguientes: (véase figura 5)
Fuente: Bly RA, Bhrany AD, Murakami CS, Sie KC. Microtia Reconstruction. Facial Plast Surg Clin North Am. 2016;24(4):577-591. doi: 10.1016/j.fsc.2016.06.011. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5950715/
1. Una porción de cartílago costal autólogo colocado por vía subcutánea.
2. Material artificial implantado que incluye un implante de polietileno poroso colocado por vía subcutánea o bajo un colgajo fascial vascularizado e injerto de piel.
3. Una prótesis de oreja adherida a la piel con un adhesivo médico o mediante implantes osteointegrados.
El manejo de la atresia aural incluye los implantes de conducción ósea, implantes de oído medio y reconstrucción quirúrgica (5, 8). Entre estas opciones, la canaloplastia tiene la ventaja de reconstruir el conducto auditivo externo y reducir la necesidad de dispositivos auditivos (8).
En cuanto a los implantes de conducción ósea, está el BAHA (Bone Anchored Hearing Aid), Bonebridge y Sophono.
Las indicaciones del BAHA son los siguientes:
Pacientes mayores de cinco años con disgenesia auditiva unilateral o bilateral que presentan hipoacusia conductiva o mixta con una vía ósea por encima de los 45 db, que no pueden emplear un audífono por vía aérea pueden ser candidatos para el mismo.
Las prótesis deben plantearse desde los 3-4 meses si la hipoacusia es bilateral. Cuando el conducto auditivo externo es permeable al menos en un lado, se propone una prótesis por vía aérea (19).
El Bonebridge utiliza un sistema de conducción ósea estimulando a través de la vibración del cráneo la cóclea en forma directa (19). Los componentes internos son una bobina receptora unida al transductor de masa flotante de conducción ósea (20).
Las indicaciones son las mismas que para el dispositivo BAHA® (19)
Las indicaciones de la cirugía funcional son los siguientes:
En caso de microtia unilateral, la mayoría de los autores coinciden en no proponer una rehabilitación auditiva quirúrgica, debido a los riesgos quirúrgicos (laberintización, parálisis facial, estenosis del conducto) y los resultados inconstantes de ésta (audición insuficiente en al menos 66 % de los casos).
Se indica una intervención quirúrgica funcional cuando la microtia es bilateral a partir de los cinco años. En caso contrario, puede proponerse una prótesis osteointegrada (25).
En la atresia aural, la corrección quirúrgica, a menudo, no es el tratamiento preferido; el resultado de la audición no es mejor que el de los dispositivos de conducción ósea, y la cirugía puede estar asociada con recurrencia o complicaciones como la estenosis del meato (31).
Con el implante de Sophono, existe la posibilidad de acoplamiento del procesador en cuanto la piel de la herida quirúrgica cicatriza por completo, en un máximo de tres o custro semanas; el implante queda completamente oculto bajo la piel, genera una menor alteración estética y disminuye el riesgo de daño del implante por manipulación (33).
Los pacientes deben ser diagnosticados y tratados por un equipo multidisciplinario: médico de familia, pediatra, genetista, audiólogo pediatra, otorrinolaringólogo pediatra o un cirujano plástico pediatra, en el que se deben considerar y coordinar las opciones para la reconstrucción tanto de la audición como del pabellón auricular durante el periodo neonatal (26, 30).
CONCLUSIONES
Es necesario estudios sistematizados en Latinoamérica que determinen la prevalencia de la microtia y atresia aural congénita. Los potenciales evocados auditivos y la audiometría constituyen el examen de elección para los casos de microtia y atresia aural congénita. La corrección quirúrgica, a menudo, no es el tratamiento preferido; el resultado de la audición no es mejor que el de los dispositivos de conducción ósea. Asimismo, se debe priorizar el aspecto funcional que el estético, ya que la pérdida de la audición temprana interviene en el desarrollo del lenguaje en el niño.

