










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Neuro-Psiquiatría
versión impresa ISSN 0034-8597
Rev Neuropsiquiatr vol.78 no.1 Lima ene. 2015
Cefalea en trombocitosis esencial: reporte de un caso juvenil.
Headache in essential thrombocytosis: report of a juvenile case.
Luis Torres Ramírez1,a, Carlos Cosentino Esquerre1,a, José J. Centeno-Arispe2,b, Rafael Suarez Reyes1,a, Yesenia Núñez Coronado1,a.
1 Departamento de Enfermedades Neurodegenerativas, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
2 Instituto Nacional de Ciencias Neurológicas. Lima, Perú
a Médico neurólogo; b Médico residente de neurología.
RESUMEN
La trombocitosis esencial (TE), es una hemopatía caracterizada por un elevado número de plaquetas (>450x109L) e hiperplasia megacariocítica de la médula ósea, poco frecuente en menores de 30 años. La cefalea se encuentra en 45% de los casos, con características similares a la migraña, planteando dificultades diagnósticas. El número de plaquetas es relevante para el pronóstico (mayor riesgo de hemorragias o eventos isquémicos) y el tratamiento sintomático de la cefalea (contraindicación de ácido acetilsalicílico en pacientes con recuento plaquetario mayor a 1 500x109L). Se reporta el caso de una mujer de 20 años de edad, que inicia hace 3 años con episodios de cefalea y conteo de plaquetas en 2195x109L, hospitalizada por reagudización de cefalea, con un recuento plaquetario de 4640x109L. Mejora con hidroxicarbamida y analgésicos parenterales remitiendo los síntomas neurológicos.
PALABRAS CLAVE: Cefalea, trombocitosis, trastorno migrañoso.
SUMMARY
Essential thrombocytosis (ET) is a blood disorder characterized by an elevated platelet count (> 450x109L) and megakaryocytic hyperplasia of the bone marrow is rare in people under 30 years. Headache occurs in 45% of cases, with migraineous characteristics, leading to difficulties in diagnosis. The number of platelets is relevant for prognosis-higher numbers correlate with more risk of bleeding or ischemic events, and guides the treatment of headaches, since the use of aspirin is contraindicated in patients with a platelet count more than 1500 x109L. We report a case of a 20 year-old woman, who presented with episodes of headache 3 years ago with a platelet count of 2195 x109L. She was hospitalized again recently for a headache exacerbation with a platelet count of 4640 x109L. After being treated with Hydroxyurea and parenteral analgesics her neurological symptoms resolved.
KEY WORDS: Headache, thrombocytosis, migraine disorders.
INTRODUCCION
La trombocitosis esencial (TE), es una hemopatía que se caracteriza por un elevado número de plaquetas circulantes e hiperplasia megacariocítica de la médula ósea. Comparte una serie de características hematológicas, clínicas y evolutivas con la leucemia mieloide crónica (LMC), la policitemia vera (PV) y la mielofibrosis idiopática (MFI), agrupándose como síndromes mielo-proliferativos crónicos (SMPC) (1). En la PV, MFI y la TE se ha identificado la presencia de la mutación V617F en el gen JAK2, presente en el 57% de casos de TE (2).
La tasa de incidencia de la TE oscila en un rango de 0,59 – 2,53x100000 habitantes, con una prevalencia de 30x100 000 habitantes, considerándose TE cuando el recuento de plaquetas supera los 450x109L (1,3-5). La TE con frecuencia se observa entre los 65 a 70 años, con un rango de edad variable, es poco frecuente en menores de 30 años y se presenta predominante en el sexo femenino (1,6,7).
Esta enfermedad suele ser asintomática en un 60% de los casos y su identificación obedece a un hallazgo durante la realización de exámenes hematológicos por otras circunstancias. Billot y colaboradores, encontraron que 29% de los pacientes con diagnóstico reciente de TE tenían manifestaciones neurológicas, las más frecuentes fueron; cefalea (45%), mareos (45%), accidente isquémico transitorio (27%); en menor frecuencia disturbios visuales, trombosis venosa, pérdida de conciencia y eventos isquémicos a nivel cerebral (1,8, 9). Por otro lado, se reportan también eventos hemorrágicos que pueden limitarse a manifestaciones cutáneas recurrentes como hematomas, epistaxis, sangrado gingival, o llegar a presentar hemorragias intracerebrales, que se observan de forma infrecuente y se presentan cuando existe alteraciones en el factor de Von Willebrand en pacientes con niveles altos de plaquetas >1500x109L (1,8, 10).
Respecto a la cefalea en pacientes con TE, no tiene características específicas pero se ha descrito como un dolor pulsátil o punzante, intermitente, de inicio repentino y de varias horas de duración, que puede ir acompañado de visión borrosa o escotomas centellantes (8,11,12), estas características semejan una migraña, planteando dificultad con su diagnóstico (13,14). Por este motivo se ha propuesto en la práctica clínica, que los pacientes que inician los episodios de cefalea en relación temporal con el diagnóstico de TE y si éstos episodios se resuelven en el espacio de tres meses de tratamiento con dosis bajas de Ácido acetil salicílico (AAS) o terapia cito-reductora, entonces puede considerarse la cefalea como manifestación de la TE (15).
El tratamiento de la cefalea en la TE, depende primariamente del control de la enfermedad de base y se puede iniciar el tratamiento con dosis bajas de ácido acetilsalicílico (AAS) que ha mostrado un nivel de eficiencia y seguridad elevado (16-18). Se debe tener en cuenta que el AAS puede desenmascarar un sangrado latente y ocasionar una hemorragia severa en pacientes con recuento de plaquetas superior a 1500x109L por alteraciones a nivel del factor de Von Willebrand, situación que contraindica su uso en estos casos(1,16).
Reporte de caso
Mujer de 20 años de edad, sin antecedentes de importancia, debuta a los 17 años con episodios de cefalea pulsátil, holocraneana, de moderada intensidad, que dura varias horas, no interfiriendo con las actividades diarias y cede con la administración de AINEs; inicialmente catalogada como parte de un proceso de infección sinusoidal, atribuido a una desviación de tabique nasal, planteándose para ello la corrección quirúrgica; al realizar exámenes de laboratorio pre-quirúrgicos se evidencia un recuento de plaquetas de 2195x109L. Se realiza estudio de medula ósea que muestra proceso mieloproliferativo, compatible con TE. La búsqueda de la mutación V617F en el gen JAK2 fue negativa. Se instauró tratamiento cito-reductor con Hidroxicarbamida, luego Interferón alfa 2b, manejando los episodios de cefalea con analgésicos no esteroideos, el paciente descontinua el tratamiento cito-reductor y controles hematológicos durante 3 años.
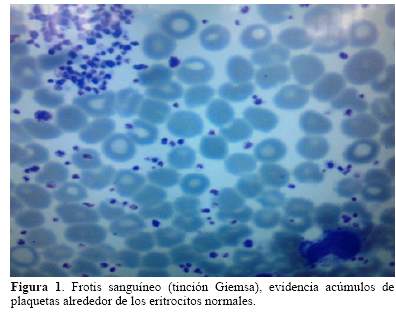
A la fecha presenta nuevo episodio de cefalea pulsátil, de inicio insidioso, intensa, acompañada de nauseas con vómitos, fotofobia, fonofobia y escotomas centellantes, los síntomas llegan a interferir las actividades diarias, al examen físico no muestra alteraciones clínicas, por lo que es catalogada inicialmente como una crisis migrañosa; durante su hospitalización se evidencia un recuento plaquetario de 4640x109L (Figura 1 y figura 2). Se indica Hidroxicarbamida 500mg cada 12 horas, tratamiento analgésico parenteral con Ketorolaco, con lo cual se logra remitir la cefalea, tras la mejoría clínica se decide su alta con analgésicos orales, cuidando de no administrar AAS por las contraindicaciones propias de esta patología.
>
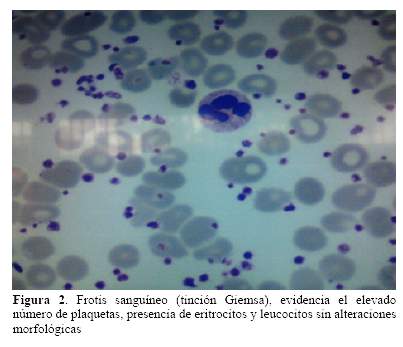
DISCUSION
El caso reportado corresponde a una mujer joven, que a los 17 años padece cuadros de cefalea recurrente y en un hallazgo de laboratorio se evidencia un recuento de plaquetas de 2195x109L; a pesar que ésta enfermedad es poco frecuente en menores de 30 años (6,7), los rangos de edad descritos son amplios e incluyen a adolescentes y jóvenes, como es el caso que presentamos. Los episodios de cefalea descritos son pulsátiles, holocraneales, de moderada intensidad y duran varias horas; características similares a otros estados patológicos, lo que hace difícil su diferenciación clínica, principalmente con la migraña, con la cual guarda una estrecha relación fisiopatológica, teniendo en cuenta que en TE hay una activación y agregación plaquetaria elevada, con la subsiguiente liberación de serotonina plaquetaria que también interviene en las alteraciones microvasculares descritas en la migraña con aura. Al respecto Frewin et. al., en una revisión hecha el 2012, menciona que desde el punto de vista práctico, la cefalea que guarda estrecha relación temporal con el diagnóstico de trombocitosis y mejora con la instauración del tratamiento cito-reductor en los primeros tres meses, generalmente corresponde a una manifestación de la TE (12). En nuestro caso la paciente posterior a su diagnóstico inicial de TE, empieza tratamiento cito-reductor (hidroxicarbamida e interferón alfa 2b), completando el estudio genético que demostró la ausencia de mutación V617F en el gen JAK2; situación relevante si se tiene en cuenta que Baxter y colaboradores, el 2005 observaron que un 57% de sus pacientes con TE presentaban una mutación puntual a nivel de este gen (2). Billot S. y col., el 2011, encontraron que el 82% los pacientes con síntomas neurológicos, tenían la mutaciónV617F en el gen JAK2, proponiendo que ésta mutación podría ser un factor de riesgo para desarrollar síntomas neurológicos en pacientes con TE, debiendo ser confirmado por futuros estudios controlados (8). En nuestro caso no se evidenció una mutación a éste nivel, pero esta situación no descarta que el origen de los síntomas neurológicos sea ocasionado por el trastorno hematológico de fondo.
En el episodio actual, es llamativo el elevado número de plaquetas que presenta nuestro caso, desde su diagnóstico hasta la fecha, si consideramos que en la serie estudiada por U. Buddeen 1993, el valor más alto de plaquetas descrito en sus pacientes no supera los 1500x109L (11). Beatrice JM., reportó un caso el 2013 en Brasil, de un niño de 8 años que inicia con cefalea y un recuento de plaquetas de 1609x109L (6). No existe una clara evidencia que correlacione la presencia o intensidad de la cefalea con el recuento de plaquetas en pacientes con TE; pero sí debe considerarse como un factor de riesgo para desarrollar eventos isquémicos a nivel cerebral, más aún si el recuento de plaquetas supera los 1500x109L, hecho que puede alterar el factor de Von Willebrand y se relaciona con manifestaciones hemorrágicas en los pacientes con TE (9-11). Nuestro caso presentó un recuento de plaquetas de 4640x109L, situación que conlleva el riesgo de eventos isquémicos e incrementa la posibilidad de una hemorragia intracerebral; además en relación al tratamiento de la cefalea se debe tener presente que el uso de AAS está contraindicada en esta situación por el mayor riesgo de sangrado (11,16-18).
Finalmente, en lo concerniente al manejo, en el episodio inicial se instauro terapia cito-reductora (hidroxicarbamida e Interferón alfa 2b) y los episodios de cefalea se controlaron con analgésicos orales (paracetamol, ibuprofeno, AAS), pero existe un periodo de tiempo en el cual no se realizó el control de la enfermedad hematológica, reagudizandose el cuadro de cefalea que propicio su internamiento actual, donde se evidencia un recuento plaquetario significativamente alto. Por el antecedente hematológico y el recuento plaquetario se retoma la terapia cito-reductora, y se indica analgésico parenteral (Ketorolaco) que permite reducir la intensidad de la cefalea hasta el alta, y el posterior manejo ambulatorio con analgésicos orales (paracetamol, ibuprofeno); derivando a la paciente a un servicio de hematología para continuar el manejo del cuadro hematológico, y normalizar el recuento plaquetario que permitirá el control de los síntomas neurológicos (18).
En conclusión, el caso presentado ilustra un diagnóstico diferencial de cefalea en pacientes jóvenes asociado a una patología hematológica, que clínicamente puede ser catalogada como cuadros de migraña, pero debe ser considerada en pacientes que inician los episodios de cefalea en relación temporal con el diagnóstico de TE y si éstos episodios se resuelven con la terapia cito-reductora, y para su manejo requiere tener en cuenta tres aspectos importantes: i) el riesgo de eventos isquémicos a nivel cerebral que presentan estos pacientes, ii) el elevado riesgo de hemorragia cerebral, propio de los pacientes con TE, que se puede incrementar con la administración de AAS en presencia de recuentos plaquetarios superiores a 1500 x 109L, y iii) el manejo de la patología de base necesario para el adecuado control de la cefalea.
REFERENCIAS BIBLIOGRAFICAS
1. Briere JB. Essential thrombocythemia. Orphanet J Rare Dis. 2007; 2:3. [ Links ]
2. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005; 365(9464):1054-61. [ Links ]
3. Spivak JL, Silver RT. The revised World Health Organization diagnostic criteria for polycythemia vera, essential thrombocytosis, and primary myelofibrosis: an alternative proposal. Blood. 2008; 112(2):231-9. [ Links ]
4. Girodon F, Bonicelli G, Schaeffer C, Mounier M, Carillo S, Lafon I, et al. Significant increase in the apparent incidence of essential thrombocythemia related to new WHO diagnostic criteria: a population- based study. Haematologica. 2009;94(6):865-9. [ Links ]
5. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002; 100(7):2292-302. [ Links ]
6. Beatrice JM, Garanito MP. Essential thrombocythemia: a rare disease in childhood. Rev Bras Hematol E Hemoter. 2013; 35(4):287-9. [ Links ]
7. Barbui T. How to manage children and young adults with myeloproliferative neoplasms. Leukemia. 2012; 26(7):1452-7. [ Links ]
8. Billot S, Kouroupi EG, Guilloux JL, Cassinat B, Jardin C, Laperche T, et al. Neurological disorders in essential thrombocythemia. Haematologica. 2011; 96(12):1866-9. [ Links ]
9. Pósfai É, Marton I, Szoke A, Borbényi Z, Vécsei L, Csomor A, et al. Stroke in essential thrombocythemia. J Neurol Sci. 2014; 336(1-2):260-2. [ Links ]
10. Adam R, Priglinger M, Harrington T, Gottlieb D, Krause M. An Unusual Cause of Cerebellar Hemorrhage in a Young Patient: Essential Thrombocythemia. Journal of Stroke and Cerebrovascular Diseases. 2014; 23(5):e373-4
11. Budde U, Scharf RE, Franke P, Hartmann-Budde K, Dent J, Ruggeri ZM. Elevated platelet count as a cause of abnormal von Willebrand factor multimer distribution in plasma. Blood. 1993; 82(6):1749-57. [ Links ]
12. Koudstaal PJ, Koudstaal A. Neurologic and visual symptoms in essential thrombocythemia: efficacy of low-dose aspirin. Semin Thromb Hemost. 1997; 23(4):365-70. [ Links ]
13. Danese E, Montagnana M, Lippi G. Platelets and migraine. Thrombosis Research. 2014; 134(1):17-22. [ Links ]
14. Borgdorff P, Tangelder GJ. Migraine: Possible Role of Shear-Induced Platelet Aggregation With Serotonin Release. Headache: The Journal of Head and Face Pain. 2012; 52(8):1298-318. [ Links ]
15. Frewin R, Dowson A. Headache in essential thrombocythaemia. Int J Clin Pract. 2012; 66(10):976- 83
16. Michiels JJ, Ten Kate FW, Koudstaal PJ, Van Genderen PJ. Aspirin responsive platelet thrombophilia in essential thrombocythemia and polycythemia vera. World J Hematol. (Internet) 2013; (Citado el 28 de noviembre de 2013) 2(2): 20- 43. Disponible en: http://www.wjgnet.com/2218-6204/full/v2/i2/20.htm
17. Michiels J. Platelet-mediated microvascular inflammation and thrombosis in thrombocythemia vera: a distinct aspirin-responsive arterial thrombophilia, which transforms into a bleeding diathesis at increasing platelet counts. Pathol Biol. 2003; 51(3):167-75. [ Links ]
18. Beer PA, Green AR. Pathogenesis and management of essential thrombocythemia. Hematology Am Soc Hematol Educ Program. 2009;621-8. [ Links ]
Correspondencia
Luis Torres Ramírez.
Jr. Ancash 1271 Lima 1 Perú.
Teléfono: 51-4117700 – Anexo 232
Correo electrónico: torresl@terra.com.pe - torresramirezl@hotmail.com
Recibido: 08/06/2014
Aceptado: 16/03/2015

