










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Neuro-Psiquiatría
Print version ISSN 0034-8597
Rev Neuropsiquiatr vol.78 no.2 Lima Apr./Jun. 2015
Síndrome de West,experiencia con una serie de casos con acceso al tratamiento de primera línea, en Lima
West Syndrome, experience in a case series with accessto first line medication, in Lima
Daniel Guillén Pinto 1,a,b, Daniel Guillén Mendoza c
1 Facultad de Medicina Alberto Hurtado, Universidad Peruana Cayetano Heredia. Lima, Perú.
a Docente; b Neurólogo Pediatra ; c Médico
RESUMEN
Objetivos: Describirlas características clínicas de una serie de pacientes con Síndrome de West(SW) con acceso a la medicación de primera línea. Material y métodos: Estudio retrospectivo observacional, de niños con SW que fueron atendidos entre 1996 y2014. Resultados: Se incluyeron 37 casos, con una promedio de inicio de espasmos de 6,4 meses, con predominio del sexo masculino (75,7%), la mayoría procedentes de Lima. La etiología más frecuente fue secundaria (83,8%),como prenatales, malformaciones cerebrales, Esclerosis Tuberosa, Síndrome de Down y causas perinatales. El SW fue controlado en 67,6% de los casos; Vigabatrina o ACTH fueron efectivas en 20/32 (62,5%). Dos pacientes fueron controlados con levetiracetam, uno con topiramato, uno con lamotrigina, y uno con cirugía. El patrón electroencefalográfico de hipsarritmia fue predominante 24/37 (64,9%). La comorbilidad neurológica fue muy frecuente (97.3%) y fue degrado leve sólo en 7/37 (18,9%). Dos pacientes fallecieron. Conclusiones: En esta serie el SW fue de causa secundaria y se controló eficientemente con vigabatrina o ACTH, por tanto, se recomienda incluir estos medicamentos en el petitorio nacional.
PALABRAS CLAVE:Síndrome de West, niños, epilepsia, vigabatrina,ACTH, levetiracetam, Perú.
SUMMARY
Objectives: To describe the clinical characteristics of a seriesof patients with West Syndrome (WS) with access to first line medication. Material and Methods: Retrospective observational study of children treated between 1996 and 2014. Results: 37 cases were included, with an average starting age of 6.4 months, predominantly males (75.7%), the majority was from Lima. The most common etiology was secondary (83.8%), as prenatal, brain malformations, Tuberous Sclerosis, Down syndrome and perinatal causes. The WS was controlled in 67.6% of the cases; Vigabatrinand ACTH were effective in 20/32 (62.5%). Two patients were controlled with Levetiracetam, one with Topitamate, one with Lamotrigine and one with surgery. Hypsarrhythmia was the predominant electroencephalographic pattern 23/37 (64.9%). Neurologic comorbidities were very frequent (97.3%) and they were mild only in 7/37 (18.9%). Two patients passed away. Conclusions: In this series, the WS had a secondary cause and was efficiently controlled with Vigabatrin or ACTH, therefore, we recommend their inclusion in the national request.
KEY WORDS: West syndrome, children, epilepsy, vigabatrine, ACTH, levetiracetam,Peru
INTRODUCCIÓN
William J. West (1793 – 1848) describió por primera vez esta enfermedad en su hijo de 4 meses, experiencia que fue publicada en 1841 (1). En la actualidad se considera que el Síndrome de West (SW) es una severa encefalopatía epiléptica que se presenta entre los 3 y 10 meses de edad, caracterizada por una tríada clínica de crisis epilépticas tipo espasmos, patrón electroencefalográficode hipsarrítmia o de salva supresión y deterioro enel desarrollo psicomotor (2-4).
La incidencia yprevalencia del SW varía de acuerdo con la región geográfica. Nelson en 1972 en EEUU, observó que afectaba a 1 de cada 4000 a 6000 nacidos vivos (5). Cowan en 1991, en una revisión epidemiológica, estimó una incidencia de 0.25 a 0.42 por cada 1000 nacidos vivos y una prevalencia de 0.14a 0.19 por 1000 (4). En un estudio más reciente, Hino-Fukuyo, en Japón, en el 2009, encontró una prevalencia de 1 por cada 2380 nacidos vivos(6). Recientemente Guillén et al, en un estudio multicéntrico en Lima, estimaron una incidencia de 1 por cada 2640 nacidos vivos (0,38 /1000)(7).
El SW se clasifica en sintomático o secundario, criptogénico e idiopático (8), siendo más frecuentes las causas secundarias, las que varían entre el 45,7% y 67% (9-11), de éstas, las más comunes son las causas prenatales (22,3 – 42,8%) (9). En el estudio multicéntrico en Lima las causas secundarias correspondieron al 36,7%, entre las que predominaron las malformaciones cerebrales y la Esclerosis Tuberosa (7).
Desde el punto de vista clínico, los espasmos son crisis generalizadas de tipo mioclónico, se presentan en salvas de decenas a centenas, la mayoría en flexión, simulando "abrazos" seguidos de irritabilidad, motivo por el cual son confundidos como cólicos del lactante (1). En estos niños el desarrollo psicomotor tiende a agravarse a medida que aumentan los espasmos constituyendo el estado encefalopático (12-14).
No existe tratamiento específico para el SW; sin embargo, el ACTH y la Vigabatrina son las drogas que han dado los mejores resultados (15-24,34). Lamentablemente estos medicamentos no se encuentran en el petitorio nacional. El objetivo del presente estudio es presentar una serie de casos de SW con mayor acceso al tratamiento recomendado, haciendo énfasis en el tratamiento y pronóstico a corto y largo plazo.
MATERIAL Y MÉTODOS
Se trata de un estudio tipo serie de casos, observacional y descriptivo, de pacientes atendidos en forma privada en Lima. Los datos se extrajeron de las historias y registros clínicos de todos los pacientes atendidos desde 1996 hasta 2014.
Todos los casos incluidos fueron diagnosticados de SW por un especialista en Neuropediatría, a través de la evaluación clínica, neuroimagen y electroencefalografía. Se excluyeron los casos en los que el paciente acudió una sola vez a consulta o si los datos encontrados en la historia clínica eran insuficientes.
Se utilizó una ficha clínica de recolección de datos, donde se registró la información acerca de las características clínicas y epidemiológicas. En concordancia con los aspectos éticos se reservó la identidad de los pacientes usando códigos de referencia.
La base de datos fue analizada haciendo uso del programa SPSS v17.0. En los casos en los que la distribución de las variables a comparar era normal se usó la prueba de T de student.
En casos de variables sin distribución normal se usaron pruebas no paramétricas.Se comparó también la efectividad de los tratamientos de primera línea en función al control de crisis con la prueba de chi cuadrado con corrección de Yates.
RESULTADOS
Fueron incluidos 37pacientes. EL 75,7% fueron varones. Veintisiete por ciento fueron referidos de provincia. El 78,4% tuvo un seguimiento promedio de 4,9 años desde su diagnóstico (6m – 19 años). Ocho pacientes abandonaron el seguimiento entre los6 y 24 meses de la primera atención (Tabla 1).
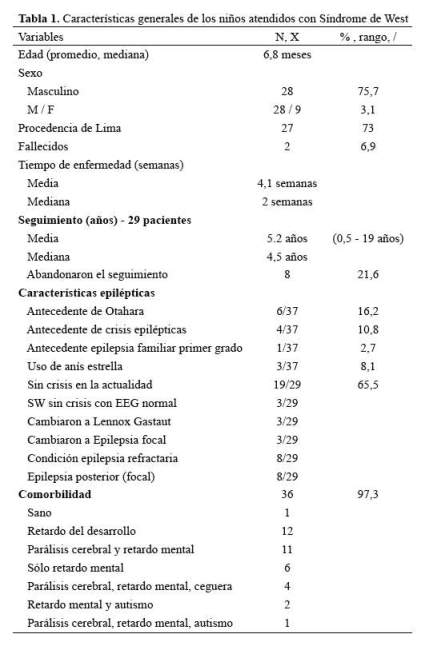
La edad promedio de inicio de los espasmos fue 6,8 meses, promedio que disminuiría a 6,4 meses al corregir la edad para los prematuros. La primera consulta se realizó con un tiempo de enfermedad promedio de 4,1 semanas y una mediana de 2 semanas (3 días– 5 meses). El antecedente de Síndrome de Otahara se observó en el 16,2% de los casos. Seis pacientes (20,7%) migraron a otra forma epiléptica y siete pacientes (24,1%) presentaron crisis focales después de un periodo asintomático (Tabla 1).
La etiología más frecuente fue secundaria 31/37 (83,8%), siendo predominantes las causas prenatales 22/37 (59,5%), en este grupo destacaron las malformaciones cerebrales 13/37 (35,1%) y la Esclerosis Tuberosa 4/37 (10,8%). Entre las causas perinatales 7/37 (18,9%) la Leucomalacia periventricular del prematuro fue la más frecuente 4/37(13.2%), y entre las postnatales las únicas registradas fueron dos casos de encefalitis, una por herpes 2 y otra por influenza (Tabla 2).

La mayoría de pacientes recibió politerapia 23/37 (62,2%), promedio de dos a tres drogas, con rango de una a seis probadas sin éxito. La droga más usada fue el ácido valproico 32/37 (86,5%), seguidadel levetiracetam 15/37 (40,5%). Con valproato como monoterapia sólo un paciente quedó libre de crisis (Tabla 3).
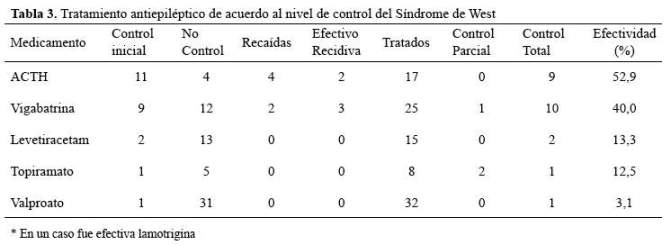
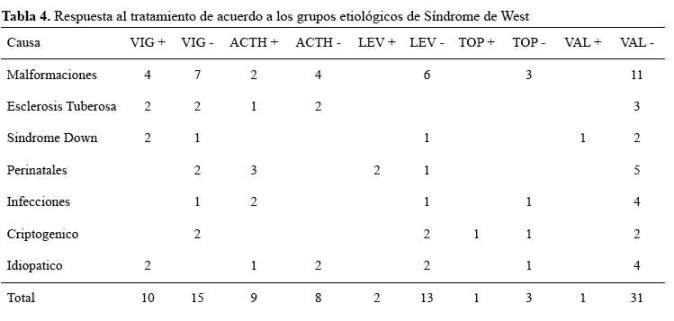
El SW fue controlado en el 67.6% de los casos, 24 casos con tratamiento medicamentoso y uno con cirugía. En 32/37 (86,5%) casos se pudo tratar con vigabatrina y/o ACTH. Vigabatrina o ACTH fueron efectivas en 20/32 (62,5%) casos. Con vigabatrina se controlaron 10 casos 10/24 (41,7%) y con ACTH 9/17 (52,9%). De 9 casos que respondieron inicialmente a vigabatrina recidivaron 2 casos, que posteriormente mejoraron con ACTH.De 11 casos que respondieron inicialmente a ACTH recidivaron5 pacientes, tres de ellos controlaron posteriormente con vigabatrina,y uno con la asociación de valproato más la motrigina. En 4 pacientes no fue efectivo el uso sucesivo de ACTH y vigabatrina, tres de ellos fueronsecundarios a malformaciones cerebrales (Tabla 3). En la comparación de ACTH y Vigabatrina contra el resto de medicamentos enfunción al control de las crisis se encontró un chi cuadrado de 23,393, con un p= 0,0001.
En 8/18 (44,4%) casos del grupo prenatales, malformaciones cerebrales, Esclerosis Tuberosa y Síndrome de Down, que se usó la vigabatrina controlaron totalmente los espasmos, contra 3/9 (33,3%) de casos que se usó el ACTH. En 2/7 casos del grupo perinatales, infecciones, idiopáticos y criptogénicos que usaron la vigabatrina controlaron los espasmos, contra 6/8 casos que usaron ACTH a dosis de 60 a 80 UI por día por dos semanas(Tabla 4).
A través del seguimiento 19/29 (65,5%) se encontraron sin crisis, 8 presentaron crisis focales entre 1 y 13 años despuésde controlar el SW. Dos fallecieron. Ocho continúan con crisis periódicas, 3 de ellos con Síndrome de Lennox Gastaut,3 con epilepsia focal refractaria y 2 con espasmos. En 24/37 (64,9%) el EEG fue de patrón hipsarritmia, sin predominio por la etiología, 18/24 con patrón hipsarritmia controlaron el SW (Tabla 1).
La comorbilidad neurológica 36/37 (97,3) más frecuente fue el retardo del desarrollo 12/37(32,4%), seguido de la parálisis cerebral más retardo mental 11/37 (29,7%),sólo uno de ellos fue de grado leve, seguido de retardo mental en 6/37 (16,2%) niños, todos de grado leve. Seis casos presentaron acidosis tubular renal (Tabla 1).
DISCUSIÓN
El SW es la encefalopatía epiléptica más frecuente y severa de la infancia, conjuntamente con el Síndrome de Ohtahara y el Síndrome de Lennox Gastaut, constituyen las encefalopatías epilépticas dependientes de la edad, que se caracterizan por un tipo de crisis epilépticas muy frecuentes y a menudo refractarias al tratamiento, EEG con patrones específicos y compromiso del desarrollo (25-28).
En el 2012 se reportó un estudio multicéntrico de SW en Lima (7), que hizo énfasis en la frecuencia y causalidad, sin embargo, pone en evidencia las dificultades del tratamiento en los pacientes. En el presente estudio se describe la experiencia con una serie de casos que tuvieron mayor acceso a los exámenes de diagnóstico,a los medicamentos de primera línea y a un seguimiento prolongado. Los padres de estos niños tuvieron un rol fundamental al identificar prontamente los espasmos infantiles como signos anormales y consultar de inmediato a su médicos, quienes también fueron eficientes en reconocer la enfermedad epiléptica.
Se presenta la experiencia de 18 años en la atención de niños con SW. La edad de presentación se encontró dentro de losrangos reportados y la etiología fue variada, entre las que destacaron lascausas prenatales, tales como las malformaciones cerebrales, Esclerosis Tuberosa, Síndrome de Down y las causas perinatales tal como se ha reportado previamente (10,11,29,30). Se encontró una mayor frecuencia de causalidad secundaria en comparación posiblemente por el mayor acceso de esta población a las pruebas diagnósticas.
No se conoce el origen del desorden epileptógeno del SW, sin embargo se entiende que diferentespatologías pueden desencadenarlo y se ha descrito la hipótesis que habría víascomunes para originar el desorden independientemente de la causa (27,28).
La gran mayoría de los pacientes de esta serie presentaron espasmos en flexión. Al respecto, en años recientes se ha destacado la utilidad de los videos para identificar el tipo y frecuencia de las crisis epilépticas, que tienden a ser de decenas de espasmos que se presentan varias veces al día en forma de salvas, de preferencia cuando despierta el niño.
Últimamente se ha descrito la importancia de hacer el diagnóstico y tratar oportunamente el SW durante las dos primeras semanas desde el inicio de las crisis. El promedio observado en este trabajofue 4 semanas con una mediana de dos, la que tendió a disminuir en los últimos años tal vez por el mayor acceso a la información de salud. Entre las recomendaciones del Subcomité para tratamiento de los espasmos infantiles de la Academia Americana de Neurología se recomienda un tiempo más corto para el tratamiento de los espasmos infantiles ya sea con terapia hormonal o VGB ya que podrían mejorar los resultados cognitivos a largo plazo (33, 34).
La eficiencia del tratamiento se encontró entre los límites reportados en la literatura (66,7%) (15-24, 34). Para iniciar oportunamente el tratamiento, los padres consiguieron los medicamentos de primera línea de otras familias con niños enfermos de SW o importándolos directamente del extranjero con el permiso de las autoridades, ya que no se encuentran en el petitorio nacional.
Podemos afirmar, con la experiencia recogida, que la vigabatrina y el ACTH fueron los medicamentos más eficaces para controlar los espasmos, tal como se reporta en la literatura(15-24). Fue interesante observar el efecto benéfico de levetiracetam para dos casos de niños prematuros con SW por leucomalacia periventricular y del topiramato para un niño con SW criptogenico. Todo lo contrario, valproato, que fue el medicamento más usado, tuvo una mínima posibilidad de éxito, la explicación para su uso fue la menor disponibilidad de otras drogas en los años 90 y principios del 2000 (31,32).
Tenemos particular comentario con respecto a la relación de la eficacia dependiendo de la etiología del SW. La vigabatrina fue más efectiva en las enfermedades prenatales, tales como malformaciones cerebrales, Esclerosis Tuberosa ySíndrome de Down. Para Esclerosis Tuberosa es conocida la indicación desde hace varios años (15, 20, 21-24) sin efectos adversos. Para el grupo de enfermedades perinatales, post infecciosas, idiopáticas y criptogénicas, la experiencia con ACTH ha sido mejor que con vigabatrina, lo cual haría suponer en un posible mecanismo inflamatorio, que esperamos se investigue en el futuro.
Muchos pacientes con SW evolucionan a otro tipo de epilepsia o encefalopatía epiléptica, como el Síndrome Lennox Gastaut. Rantala describe que 10 al 25% evolucionaron a Síndrome Lennox Gastaut; este síndrome se caracteriza por un tipo especial de epilepsia y un patrón electroencefalográfico de polipuntas y punta onda a 2,5 ciclos por segundo (16). En esta serie se describen tres niños, con una proporción semejante a la reportada. Con una mayor número de casos faltaría por determinar si la demora en el tratamiento o la etiología subyacente tiene un efecto directo sobre su frecuencia.
Pese a que la limitante del estudio fue su referencia a un grupo de pacientes con mayor acceso a los recursos de tratamiento, con su presentación hemos querido describir que los niños con SW en nuestro medio tienen diagnósticos similares al resto del mundo y por tanto pueden responder con las mismas expectativas, a pesar de ser una de la patologías más severas de la infancia.
En conclusión, la atención y manejo del SW ennuestro medio es un reto para los especialistas en Neuropediatría. La serie presentada describe la eficacia del tratamiento recomendado con ACTH y vigabatrina, abre algunas expectativas sobre el uso de levetiracetam, topiramato y el rol de la cirugía de epilepsia.
REFERENCIAS BIBLIOGRAFICAS
1.West WJ. On a peculiar form of infantile convulsions. Lancet. 1841; 1:724. [ Links ]
2.Trevathan E, Murphy CC, Yeargin-Allsopp M. The descriptive epidemiology of infantile spasms among Atlanta children. Epilepsia. 1999; 40:748. [ Links ]
3.Kurokawa T, Goya N, Fukuyama Y, Susuki M, Seki T, Ohtahara S. West syndrome and Lennox-Gastaut syndrome: a survery of natural history. Pediatrics. 1980; 65: 81-8. [ Links ]
4.Hudson LS. The epidemiology and natural history of infantile spasms. J Child Neurol. 1991;6(4):355-64. [ Links ]
5.Nelson K. Discussion. In: Alter M, Hauser WA. (eds). The epidemiology of epilepsy: a workshop. Washington DC: US Government Printing Office; 1972. p. 78. [ Links ]
6.Hino-Fukuyo N, Haginoya K, Iinuma K, Uematsu M, Tsuchiya S. Neuroepidemiology of West syndrome and early infantileepileptic encephalopathy in Miyagi Prefecture, Japan. Epilepsy Res. 2009; 87(2-3):299- 301. [ Links ]
7. Guillén D, Sánchez JP, Koc D, et al. Síndrome West: Estudio multicéntrico en Lima. Rev Peru Pediatr. 2012; 65(2): 81-88. [ Links ]
8.Brna PM, Gordon KE, Dooley JM, Wood EP. The epidemiology of infantile spasms. Can J Neurol Sci. 2001; 28:309. [ Links ]
9.Commission on Pediatric Epilepsy of the International League Against Epilepsy. Workshop on infantile spasms. Epilepsia. 1992; 33:195. [ Links ]
10.Watanabe K. West syndrome: etiological and prognostic aspects. Brain Dev. 1998;20: 1. [ Links ]
11.Ohtahara S, Ohtsuka Y. Prenatal etiologies of West syndrome. Epilepsia. 1993; 34: 716-22. [ Links ]
12.Riikonen R. A long-term follow-up study of 214 children with the syndrome of infantile spasms. Neuropediatrics.1982; 13:14-23. [ Links ]
13.Bombardieri R, Pinci M. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. 2010;14(2):146-9. [ Links ]
14.Goh S, Kwiatkowski DJ, Dorer DJ, Thiele EA. Infantile spasms and intellectual outcomes in children with tuberous sclerosis complex. Neurology. 2005; 65:235. [ Links ]
15.Mackay MT, Weiss SK, Adams-Webber T, et al. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology. 2004; 62(10):1668-81. [ Links ]
16.Baram TZ, Mitchell WG, Tournay A, Snead OC, Hanson RA, Horton EJ. High-dose corticotrophin (ACTH) versus prednisone for infantile spasms: a prospective, randomized, blinded study. Pediatrics. 1996; 97 (3): 375-9. [ Links ]
17.Ibrahim S, Gulab S, Ishaque S, Saleem T. Clinical profile and treatment of infantile spasms using vigabatrin and ACTH - a developing country perspective. BMC Pediatrics. 2010; 10:1-9. [ Links ]
18.Zupanc ML. Infantile spasms. Expert Opin Pharmacother.2003; 4(11): 2039-48. [ Links ]
19.Riikonen R. The latest on infantile spasms. Curr Opin Neurol. 2005; 18(2):91-5. [ Links ]
20.Hancock EC, Osborne JP, Edwards SW. Treatment of infantile spasms. Cochrane Database Syst Rev. 2008; 4:CD001770. [ Links ]
21.Gaily E. Vigabatrin monotherapy for infantile spasms. Expert Rev Neurother. 2012;12(3):275-86. [ Links ]
22.Willmore LJ, Abelson MB, Ben-Menachem E, Pellock JM, Shields WD. Vigabatrin: 2008 update. Epilepsia. 2009; 50(2):163-73. [ Links ]
23. Fejerman N, Cersósimo R, Caraballo R, et al. Vigabatrinas a first-choice drug in the treatment of West syndrome. Child Neurol. 2000; 15(3):161-5. [ Links ]
24.Vigevano F, Cilio MR. Vigabatrin versus ACTH as first-line treatment for infantile spasms: a randomized, prospectivestudy. Epilepsia. 1997; 38:1270. [ Links ]
25.Go CY, Mackay MT, Weiss SK, et al. Evidence-based guideline update: Medical treatment of infantile spasms: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2012; 78:1974-1980. [ Links ]
26.Vigevano F, Fusco L. The idiopathic form of West syndrome. Epilepsia.1993; 34:743-46. [ Links ]
27. Henriques-Souza AM, Ataide JL, Gomes S. Vigabatrina no tratamento da Síndrome de West avaliação clínica e eletrencefalográfica em 13 pacientes. Arq Neuropsiquiatr. 2007; 65(1): 144-149. [ Links ]
28. Pozo A, Pozo D. Síndrome de West, etiología, fisiopatología: aspectos clínicos y pronósticos. Rev Cubana Pediatr. 2002; 74(2):151-61. [ Links ]
29.Rantala H, Putkonen T. Occurrence, outcome, and prognostic factors of infantile spasms and Lennox-Gastaut syndrome. Epilepsia. 1999; 40:286. [ Links ]
30.Cusmai R, Ricci S, Pinard JM. West syndrome due to perinatal insults. Epilepsia. 1993; 34: 738-42. [ Links ]
31. Goldberg-Stern H, Strawsburg RH, Patterson B, et al. Seizure frequency and characteristics in children with Down syndrome. Brain Dev. 2001; 23: 375. [ Links ]
32.Napuri S, Gall E, Dulac O, Chaperon J, Riou F. Factors associated with treatment lag in infantile spasms. Dev Med Child Neurol. 2010; 52 (12): 1164-6. [ Links ]
33.Glauser TA, Clark PO, McGee K. Long-term response to topiramate in patients with West syndrome. Epilepsia. 2000; 41(S1): S91. [ Links ]
34.Dyken PR. Short term effects of valproateon infantile spasms. Pediatr Neurol. 1985; 1(1):34-7. [ Links ]
Correspondencia:
DanielGuillén Pinto
Calle Beethoven 429 San Borja, Lima, Perú
Correo electrónico:
Recibido:15/05/2015
Aceptado: 22/06/2015

