










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Neuro-Psiquiatría
Print version ISSN 0034-8597
Rev Neuropsiquiatr vol.78 no.3 Lima July 2015
Síndrome cortico-basal como una presentación clínica de una enfermedad de Creutzfeldt-Jakob esporádica.
Corticobasal syndrome as a clinical spectrum of a sporadic Creutzfeldt-Jakob disease.
Luis Torres-Ramírez 1,a, Hugo Sarapura-Castro1,b, Carlos Cosentino-Esquerre1,a,Miriam Vélez-Rojas1,a
1 Departamento de Enfermedades Neurodegenerativas, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
a Médico neurólogo; b Médico residente de neurología
RESUMEN
La enfermedad de Creutzfeldt-Jakob (ECJ) es una enfermedad neurodegenerativa caracterizada por demencia rápidamente progresiva, mioclonías, compromiso motor y alteraciones características en los exámenes auxiliares; sin embargo existen presentaciones clínicas atípicas del cuadro. Presentamos un caso de ECJ esporádica en asociación clínica con un síndrome cortico-basal caracterizado por apraxia de extremidades, déficit sensorial cortical, fenómeno del miembro ajeno, bradicinesia y rigidez asimétricos; quees la presentación clásica de la degeneración cortico-basal. Además los hallazgos en el electroencefalograma, resonancia magnética cerebral y resultado de la proteína 14-3-3en LCR fueron compatibles con ECJ esporádico probable. Este caso sugiere que el compromiso neurológico asimétrico puede asociarse a ECJ esporádico.
PALABRAS CLAVE: Degeneración cortico-basal, demencia, priones, síndrome de Creutzfeldt-Jakob.
SUMMARY
Creutzfeldt-Jakob disease (CJD) is aneuro degenerative disease characterized by rapidly progressive dementia, myoclonus, motor impairment and typical features on complementary tests; however, unusual clinical features might be associated. Wereport one case of sporadic CJD associated with corticobasal syndrome characterized by asymmetric limb apraxia,cortical sensory impairment, alien limb phenomenon, bradykinesiaand rigidity; which is the classic clinical spectrum of the corticobasaldegeneration. In addition, findings in electroencephalography (EEG), brainmagnetic resonance imaging (MRI) and positive CSF protein 14-3-3 werecompatible with probable sporadic CJD. This case suggestthat asymmetric neurologic impairment may be associated with sporadic CJD.
KEYWORDS: Corticobasal degeneration, Creutzfeldt-Jakob syndrome, dementia, prions.
INTRODUCCIÓN
La enfermedad de Creutzfeldt-Jakob (ECJ) es la enfermedad por priones más frecuente en humanos (1). En la ECJ se produce un plegamiento anormal de la proteína priónica (PrPC), dando lugar a la acumulación de la isoforma patológica (PrPSc),en cuya estructura existe predominio de láminas tipo b, lo cual la hace relativamente resistente a la degradación proteolítica en comparación a la PrPC (2). El mecanismo preciso de este mal plegamiento sigue siendo incierto. El PrPSces el principal, sino es el único, constituyente de la transmisibilidad de la enfermedad.
La presentación clínica se caracteriza por un síndrome demencial rápidamente progresivo y otros síntomas neurológicos (1). La ECJ tiene cuatro formas de presentación siendo la forma esporádica (esECJ) la más frecuente. La esECJ tiene una incidencia anual de 1-2 casos/millón a nivel mundial (3).
La degeneración cortico-basal (DCB) es una entidad neurodegenerativa que se enmarca en el subgrupo de los “Parkinsonismos-Plus”. La presentación clínica clásica de la DCB es el síndrome cortico basal (SCB) que incluye características de disfunción cortical (apraxia de extremidades y oculomotora, déficit sensorial cortical, miocloníasy el fenómeno del miembro ajeno o “alienlimb”) y disfunción de ganglios basales (bradicinesia, rigidez asimétrica progresiva, distonía, temblor). Sin embargo el SCB no es exclusivo de la DCB ya que se puede presentar en otras enfermedades neurológicas (4).
Se presenta el caso clínico de una paciente con esECJque inicia su enfermedad con un SCB.
Caso clínico
Mujer de 70 años de edad, natural y procedente de Lima, con hipertensión arterial desde hace 15 años. Comienza su enfermedad cuatro meses antes del ingreso a nuestra institución, con cuadro clínico de inicio insidioso y curso progresivo caracterizado por dificultad para hablar y caminar. Un mes antes notan lentitud en la ejecución de sus movimientos y debilidad en el hemicuerpo izquierdo, cambio en el comportamiento, tristeza constante, irritabilidad y presencia de sobresaltos musculares a nivel de extremidad superior izquierda, que se presenta “cada vez que se asusta”. Es traída a esta institución por presentar problemas en la marcha, necesitando apoyo pues se desvía a la izquierda.
Al examen clínico se la encontró en mal estado general, regular estado de nutrición, regular estado de hidratación, endecúbito dorsal pasivo. Al examen neurológico se la encuentra despierta, orientada parcialmente en tiempo, espacio y persona. Lenguaje poco fluido y claro, con iniciativa de intervenir de forma espontánea, ejecuta adecuadamente órdenes simples y con dificultad órdenes complejas, nomina objetos correctamente, disartria moderada, asterognosia, agrafestesia en el hemicuerpo izquierdo y no discrimina entre dos puntos, heminegligencia, rigidez en el hemicuerpo izquierdo con leve hemiparesia. Fenómeno del miembro ajeno en extremidad superior izquierda. Presencia de mioclonías reflejas a predominio del extremidad superior izquierda que aparecen tras estímulos táctiles y auditivos. Al ingreso en el Mini Mental Test presentó un puntaje 26/30. La evaluación neuropsicológica evidenció una apraxia ideomotora,constructiva, del vestir y bucofacial. Además un síndrome ansioso-depresivo de grado moderado.
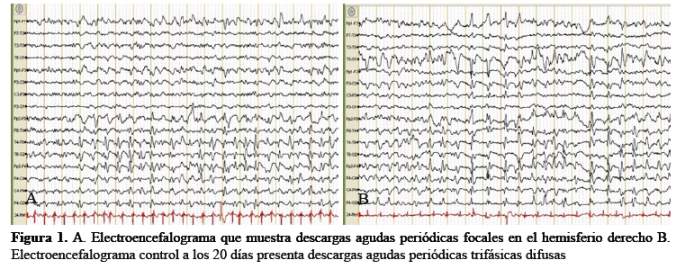
Los exámenes auxiliares que incluyeron hemograma completo y bioquímica sanguínea, perfil tiroideo y dosajede vitamina B12, fueron normales. RPR: No reactivo. El estudio de líquido cefalorraquídeo mostró 1 célula, 100% de mononucleares, 64mg/dl de glucosa, 26 mg/dl dealbúmina y cultivo para gérmenes negativo. El dosaje de la proteína 14-3-3 en líquido cefalorraquídeo fue de3,9 ng/ml (VN: 0-1,5). El electro encefalograma mostró disfunción cortico-subcortical difusa con presencia de descargas epileptiformes periódicas lateralizadas al hemisferio derecho (Figura 1 (A)), sin embargo veinte días después, las descargas se generalizaron con presencia de ondas trifásicas (Figura 1 (B)).
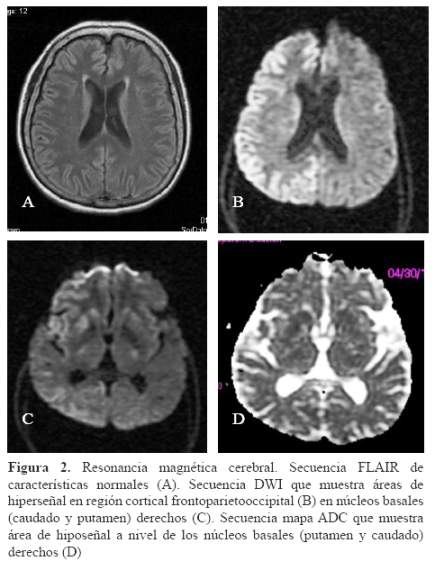
La resonancia magnética cerebral, en la secuencia de difusión (DWI), presentó hiperseñal a nivel de la región cortical y ganglios basales del hemisferio derecho e hiposeñal en el mapa ADC, a nivel de los ganglios basales que no se ven en la secuencia FLAIR (Figura 2).
Durante la evolución de la enfermedad, las mioclonías aumentaron en frecuencia y se generalizaron, mutismo aquinético y curso con una infección respiratoria intrahospitalaria. La paciente tuvo un deterioro rápidamente progresivo y fue dada de alta a solicitud de la familia.
DISCUSIÓN
La esECJ es la más frecuente de las enfermedades priónicas. Estudios previos han identificado una edad de inicio entre 55 y 75 años (promedio de 61.5 años) (5). La presentación clínica puede ser variada en el comienzo de la enfermedad, generando dificultades diagnósticas. Los síntomas iniciales en un tercio de los casos son sistémicos e inespecíficos como fatiga, trastorno del sueño e hiporexia; un tercio de pacientes se presentan con trastornos cognitivo/conductuales y el tercio final con síntomas focales como trastorno visual, ataxia cerebelosa, afasia o déficit motor. La enfermedad progresa rápidamente con deterioro cognitivo, presencia de mioclonías sensiblesal estímulo, síntomas piramidales y rigidez, y trastorno de la conducta. Las mioclonías son el síntoma más importante y está presente en casi el 90% de los casos (6).Al final del curso de la enfermedad la mayoría de pacientes van a un estado de mutismo aquinético. La muerte ocurre dentro de los 12 meses de iniciada la enfermedad en el 85% al 90% de los pacientes.
El diagnóstico posible de la esECJ, según los criterios actuales propuestos por Zerr et al.,(7), requieren la presentación de un síndrome demencial rápidamente progresivo y dos signos clínicos: mioclonías, trastornos visuales o cerebelosos, signos piramidales/extrapiramidales y mutismo aquinético, con una duración menor de dos años. Y pasa a ser probable cuando se añade al menos un hallazgo típico en las pruebas complementarias como el electroencefalograma(EEG), el análisis de líquido cefalorraquídeo (LCR) o la resonancia magnética (RM) cerebral. El EEG muestra complejos de ondas agudas periódicas bifásicas o trifásicas de 1-2ciclos/segundo, usualmente generalizadas y difusas. Sin embargo algunos pacientes presentan descargas periódicas focales asociados al cuadro clínico y hallazgos en la RM cerebral (8). Los hallazgos en el EEG se presentan sólo en el 70% de las formas esporádicas (9). El análisis de LCR con aumento de la proteína 14-3-3 representa daño neuronal; es sensible y específica para esECJ. Sin embargo baja su especificidad en eventos neurológicos agudos que causan demencia progresiva (inflamación, enfermedades cerebrovasculares, tumores del SNC, crisisepilépticas) (10). Torres et al., han publicado recientemente el primer reporte de ECJ en el Perú, en el cual se describen las características clínico-patológicas y pruebas complementarias en 11 casos (11).
LaDCB es un parkinsonismo atípico poco frecuente y su diagnósticoclínico es difícil debido a su heterogeneidad clínica. En1997 Vélez y col. presentaron dos casos de DCB describiendo características clínicas y neuro-imagenológicassimilares a las descritas en la literatura (12). El diagnóstico posiblede DCB incluye una presentación de inicio insidioso y curso progresivode un año de evolución como mínimo, sin considerar la edad de inicio, con historia familiar, con un síndrome clínico de DCB, y con una mutación genética asociada (por ejemplo MAPT). Y pasa aser probable cuando la edad de inicio es igual o mayor de 50 años y conla exclusión de historia familiar y mutación genética(13).
LaesECJ se distingue de la DCB por la duraciónmás corta de la enfermedad y las pruebas complementarias como son la presencia de la proteína 14-3-3, la presencia de los complejos de ondas agudas periódicas en el EEG y los hallazgos en la RM cerebral en FLAIRy/o DWI. Sin embargo debido a la existencia de diversos subtipos moleculares en la esECJ se puede presentar un curso clínico menos específico con características similares a otras enfermedades neurodegenerativas como la DCB. Boeve etal. en una serie de 34 casos de SCB, encontraron que 3(8%) tuvieron el diagnóstico de esECJ (14).
El fenómeno del miembro ajeno es una actividad motora autónoma, que puede ser movimientos sin propósito, compulsivos con un propósito o autodestructivos. Puede estar acompañado de síntomas motores como apraxia de extremidades, hemiparesia, hemiataxia, mioclonías reflejas corticales, distonía de acción, atetosis o hemibalismo(15), descrito en extremidades superiores. Existen diversos reportes de casos que relacionan este signo con esECJ, donde fue una de las manifestaciones iniciales del cuadro clínico antes o después de la demencia (16). A su vez; casos de esECJ definitiva, con deterioro cognitivo y fenómeno del miembro ajeno izquierdo en los cuales los exámenes complementarios como el aumento de proteína 14-3-3 y los hallazgos del EEG fueron poco contributorios.
En nuestro caso, el cuadro clínico es de inicio tardío, con síntomas motores focales y trastorno del lenguaje desde el inicio. Presentó hemiparesia, bradicinesia, rigidez y el fenómeno del miembro ajeno, todas ellas de inicio focal en el hemicuerpo izquierdo, con leve deterioro cognitivo, lo que llevo a pensar en primera instancia que se trataba de un SCB probable de corta evolución y de rápida progresión. Según los criterios diagnósticos de Armstrong et al., no podría corresponder a una DCB, ya que el tiempo de evolución es menor de un año (13). El SCB probable puede corresponder a múltiples enfermedades neurodegenerativas como parálisis supranuclear progresiva, enfermedad de Pick, demencia frontotemporal, enfermedad de Alzheimer y esECJ. En las pruebas complementarias como el EEG se presentó descargas epileptiformes periódicas focales en todo deltrazado del hemisferio derecho, que luego de 20 días se generalizan y presenta descargas periódicas agudas trifásicas. Presentó aumento de la proteína 14-3-3 y en las imágenes una restricción de la difusión con hiperseñalen la secuencia DWI en la corteza y ganglios basales, e hipo señalen la secuencia mapa ADC en los ganglios basales, del hemisferio derecho. Todo ello nos hace pensar que se trata de un cuadro clínico, electroencefalográfico y neuroimagenológico eminentemente focal con una progresión rápidamente progresiva, que correspondería a una forma clínica de presentación poco frecuente de una esECJ (17,18).
Enla DCB, las mioclonías y el fenómenodel miembro ajeno ocurren generalmente luego del primer año del iniciode la enfermedad; sin embargo aparece como síntomas tempranos y enalgunos casos como síntomas iniciales en una esECJ (19). En nuestro caso las miocloníasy el signo del miembro ajeno aparecieron a los cuatro meses del inicio de lossíntomas, aunque no fueron los primeros síntomas. Enadición a estas diferencias clínicas, la RM cerebral y, particularmente la secuencia DWI fueron útiles en la ayudadiagnóstica.
Mader et al., sugieren que se deben tener en cuenta ciertos hallazgos dentro de los criterios actuales para eldiagnóstico de esECJ como que la rápida progresión del deterioro cognitivo y las miocloníasno son exclusivas de la esECJ (20); las manifestaciones focales/lateralizadas no son típicas de esECJ, sin embargo pueden estar presentes, lo que dificultasu diagnóstico temprano; las pruebas complementarias son de gran ayuda para el diagnóstico en estadios tempranos de la enfermedad y cuando el diagnostico se encuentra incierto.
En conclusión, presentamos este caso para llamar la atención sobre las formas inusuales de presentación clínica de la esECJ, pudiéndose confundir con parkinson ismosatípicos de tipo DCB.
REFERENCIAS BIBLIOGRÁFICAS
1. Johnson RT. Priondiseases. Lancet Neurol. 2005; 4(10):635-42. [ Links ]
2. Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363-83. [ Links ]
3. Mead S, Stumpf MPH, Whitfield J, Beck JA, Poulter M, Campbell T, et al. Balancing selection at the prion protein geneconsistent with prehistoric kurulike epidemics. Science. 2003; 300(5619):640-3. [ Links ]
4. Wadia PM, Lang AE. The many faces of corticobasal degeneration. Parkinsonism Relat Disord. 2007;13(S3):S336-40. [ Links ]
5. Belay ED. Transmissible spongiform encephalopathies in humans. Annu Rev Microbiol. 1999; 53:283-314. [ Links ]
6. Kretzschmar HA, Ironside JW, DeArmond SJ, Tateishi J. Diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Arch Neurol. 1996; 53(9):913-20. [ Links ]
7. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain J Neurol. 2009;132(10): 2659-68. [ Links ]
8. Cambier DM, Kantarci K, Worrell GA, Westmoreland BF, Aksamit AJ. Lateralized and focal clinical, EEG, and FLAIR MRI abnormalities in Creutzfeldt-Jakob disease. Clin Neurophysiol. 2003;114(9):1724-8. [ Links ]
9. Sharma S, Mukherjee M, Kedage V, Muttigi MS, Rao A, Rao S. Sporadic Creutzfeldt-Jakob disease:a review. Int J Neurosci. 2009; 119(11):1981-94. [ Links ]
10. Stoeck K, Sanchez-Juan P, Gawinecka J, Green A, Ladogana A, Pocchiari M, et al.Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt-Jakob disease and rapid dementias: a longitudinal multicentre study over 10 years. Brain J Neurol. 2012; 135(10):3051-61. [ Links ]
11.Torres-Ramírez L, Ramírez-Quiñones J, Cosentino-Esquerre C, Vélez-Rojas M, Flores-Mendoza M, Rivas-Franchini D, et al. Enfermedad de Creutzfeldt-Jakob en el Perú: reporte de once casos. Rev Peru Med Exp Salud Pública. 2014; 31(2):364-9. [ Links ]
12.Velez M, Cosentino C, Torres L. Degeneración corticobasal: Reporte de dos casos. Rev Neurol. 1997; 31-3: 19-22. [ Links ]
13. Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013; 80(5):496-503. [ Links ]
14. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54 (S5):S15-9. [ Links ]
15. Hanna PA, Doody RS. Alien limb sign. Adv Neurol. 2000; 82:135-45. [ Links ]
16. Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002; 61(11):935-46. [ Links ]
17. Anschel DJ, Simon DK, Llinas R, Joseph JT. Spongiform encephalopathy mimicking corticobasal degeneration. Mov Disord. 2002; 17(3):606-7. [ Links ]
18. Moreaud O, Monavon A, Brutti-Mairesse MP, Grand S, Lebas JF. Creutzfeldt-Jakob disease mimicking corticobasal degeneration clinical and MRI data of a case. J Neurol. 2005; 252(10):1283-4. [ Links ]
19. Lee W, Simpson M, Ling H, McLean C, Collins S, Williams DR. Characterising the uncommon corticobasal syndrome presentation of sporadic Creutzfeldt-Jakob disease. Parkinsonism Relat Disord. 2013; 19(1):81-5. [ Links ]
20. Mader EC, El-Abassi R, Villemarette-Pittman NR, Santana-Gould L, Olejniczak PW, England JD. Sporadic Creutzfeldt-Jakob disease with focal findings: caveats to current diagnostic criteria. Neurol Int. 2013;5(1):1-5. [ Links ]
Fuente de financiamiento: autofinanciado.
Conflictos de interés: los autores declaran no tener ningún conflicto deinterés
Correspondencia
Elison Hugo Sarapura Castro
Correo electrónico: hugosarcas@gmail.com
Recibido:02/05/2015
Aceptado:16/08/2015
