










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Neuro-Psiquiatría
Print version ISSN 0034-8597
Rev Neuropsiquiatr vol.78 no.4 Lima Oct. 2015
Homocistinuria, una enfermedad metabólica de diagnóstico tardío en el Perú.
Homocystinuria, a metabolic disease of late diagnosis in Peru.
MarioR. Cornejo-Olivas1,a, Vilma G. Chávez-Pasco1,b, Miguel A. Inca-Martínez1,c, Diego M. Véliz-Otani1,c, Anali P. Mora-Alférez 2,b, Mario R. Velit-Salazar 1,4,d,Arantxa Sánchez-Boluarte1,4,d, Maria Quiroga de Michelena3,b, Pilar Mazzetti1,5,a
1 Centro de Investigación Básica en Neurogenética, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
2 Equipo Funcional de Genética y Biología Molecular, Instituto Nacional de Enfermedades Neoplásicas. Lima, Perú.
3 Instituto de Medicina Genética. Lima, Perú.
4 Facultad de Medicina Humana Alberto Hurtado, Universidad PeruanaCayetano Heredia. Lima,Perú.
5 Facultad de Medicina Humana, Universidad Nacional Mayor de SanMarcos. Lima, Perú.
a Médiconeurólogo ;bMédico genetista ; cBiólogo genetista ; d Estudiantede pregrado
RESUMEN
La Homocistinuria,es un desorden metabólico autosómico recesivo, cuya forma clásica es causada por deficiencia de cistationina β-sintasa, debido a mutaciones en el gen CBS (Cr21q22.3). Se describe el caso de un varón de 17 años con hipopigmentación de piel y faneras, retraso psicomotor moderado, hábito marfanoide, miopía severa, subluxación del cristalino bilateral, que además presentóeventos psicóticos y una hemiparesia izquierda secundaria a un infartolacunar. La determinación de homocisteína en plasma se encontró elevada (>9,9mg/dl), así como niveles altos denitroprusiato de sodio en orina(4+)que confirmaron el diagnóstico clínico de homocistinuria.La homocistinuria clásica genera múltiples complicaciones a nivel dérmico, oftalmológico, cognitivo,osteoarticular y psiquiátrico; quepodrían evitarse con un diagnóstico y tratamiento oportuno a través del tamizaje neonatal, aún no disponible en la mayoría de centros asistenciales en el Perú.
PALABRAS CLAVE: Enfermedades metabólicas, hábito marfanoide, homocistinuria,Perú, diagnóstico tardío.
SUMMARY
Homocystinuria is an autosomal-recessive metabolic disorder whose classical phenotype is caused by a deficiency of cystathionine β-synthase, dueto mutations within the CBS gene(Cr21q22.3). Here in we report a 17 years old man with hypopigmented skin and hair, mental retardation, marfanoid habitus, severe myopia, bilateral lens subluxation, psychotic episodes, and left-sided hemiparesis secondary to alacunar brain infarction. Laboratory tests showed increased levels of homocysteine (>9.9mg/dl) in plasma and high levels of urinary sodium nitroprusside (4+), consistent with the clinical diagnosis of classical homocystinuria. This systemic disorder includes dermal, ophthalmic, cognitive, osteoarticular and psychiatric alterations, all of which could be potentially prevented with early diagnosis and therapy as part of new born screening, which is still unavailable in Peru.
KEY WORDS: Homocystinuria, marphanoid habitus, metabolicdisorders, Peru, delayed diagnosis.
INTRODUCCIÓN
La homocistinuria es un error innato del metabolismo caracterizado por la acumulación de homocisteínaa nivel del sistema nervioso central, vascular, esquelético y ocular. La prevalencia mundial de homocistinuria se estimaen1-9/100000 habitantes, aunque se asume que esta puede estar subestimada por los casos no diagnosticados (1).
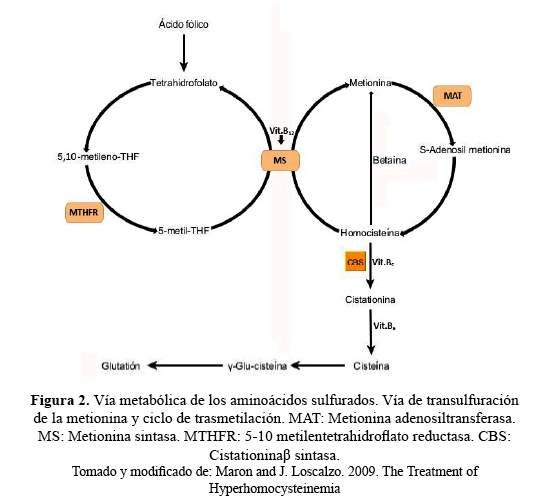
La homocisteína es un aminoácido azufrado no esencial presente en la intersección de rutas metabólicas que regulan los niveles de metionina, folato y fuentes de grupos metilo para numerosos sustratos (2). La homocistinuria puede ser causada por mutaciones en al menos cinco genes distintos: CBS, MMADHC, MTHFR, MTR y MTRR (3). La forma clásica de esta enfermedades de herencia autosómica recesiva, producida por deficiencia de la enzimacistationina β-sintasa (CBS), la cual cataliza la conversión dehomocisteína en cistationina, que forma parte del primer paso de la ruta de transulfuración de la homocisteína (4). El gen CBS (21q22.3), que codifica la cistationina beta-sintasa, registra al menos 164 mutaciones distintas (5).
La deficiencia en la función de CBS genera la acumulación de homocisteína, metionina, S-adenosilmetionina (SAM), S-adenosilhomocisteína (SAH) y sarcosina; además de la reducción de la producción decistationina y cisteína, alterando varias rutas metabólicas (6).
En Latinoamérica, existen pocas descripciones de esta enfermedad, la mayoría referidas a población pediátrica. Describimos el primer caso peruano de homocistinuria de diagnóstico en la adolescencia.
Reporte de caso
Previa autorización por el comité Institucional de Ética en Investigación del Instituto Nacional de Ciencias Neurológicas, se describe el siguiente caso. Varón de 17 años (Individuo IV 1, Figura 1 y figura 2), mestizo, hijo de padres consanguíneos, nacido de parto vaginal pretérmino de 33semanas, peso al nacer de 1,7 kg. Al examen del recién nacido se destaca la hipopigmentación de piel y faneras. Presentó retraso del desarrollo psicomotora predominio de motricidad fina y del lenguaje, bajo rendimiento escolar, solo 8 años de estudio. A los seis años fue operado por luxación bilateral de cristalino y glaucoma bilateral. A los 14 años presentó episodio brusco de hemiparesia izquierda, concomitantemente aparecieron episodios de agitación psicomotora y alucinaciones visuales, así como conductas compulsivas.
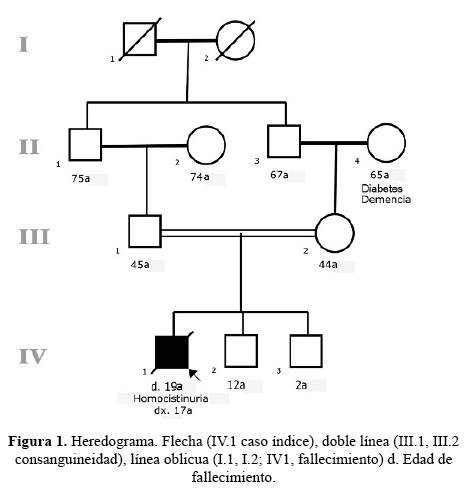
Al examen físico, realizado a los 17años, se evidenció hábito marfanoide, constitución delgada, con brazada de 1,70m, talla de 1,57m, la relación brazada/estatura: 1,1correspondiente a dolicoestenomelia. Piel clara con livedo reticularisy estrías gruesas múltiples en tórax y región inguinal, cabello hipopigmentado, fino y quebradizo con implantación capilar anterior alta y posterior baja, cejas y pestañas hipopigmentadas.
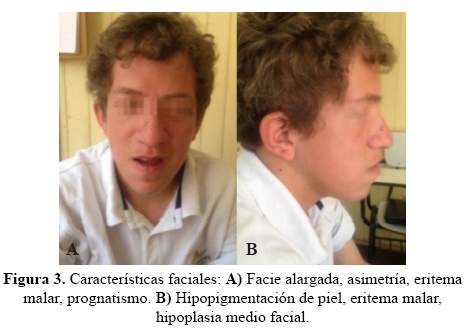
Facie alargada, asimetría, fisuras palpebrales alargadas con inclinación hacia abajo, incremento del pliegue infraorbital, engrosamiento de alas nasales, eritema malar, base nasal ancha, hipoplasia medio facial, labios engrosados, prognatismo, barbilla puntiaguda y alargada; paladar ojival, petequias en paladar blando; orejas con leve rotación posterior (Figura 3).
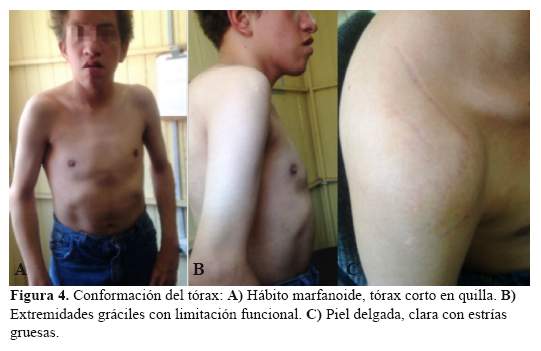
Tórax corto en quilla, escoliosis con extremidades gráciles y alargadas con limitación a la extensión en grandes articulaciones (codos, rodillas y tobillos), postura en garra (Figura 4).
Aranodactilia, ortejos y pies alargados y cavos (Figura5).
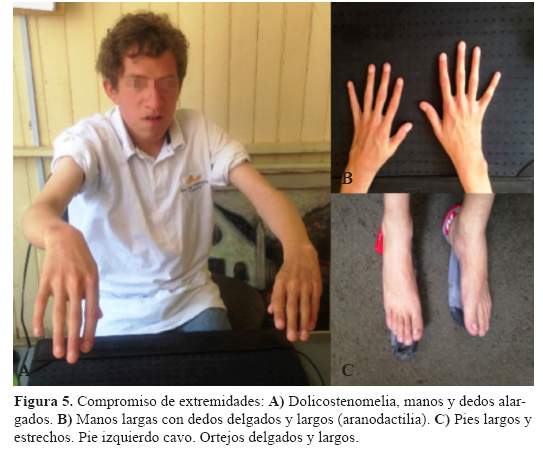
Al examen neurológico inicial, presentaba lenguaje incoherente, tics vocales tipo tos, irritabilidad con tendencia a la autoagresividad, hiperalerta con episodios de agitación psicomotriz, hemiparesia izquierda pura y proporcional con hiperreflexia, Babinski izquierdo. Espasticidad con tendencia a la flexión de miembro superior izquierdo.
Exámenes auxiliares: nitroprusiato de sodiopositivo 4+(cisteína y homocisteína) en orina. Homocisteína en sangre>9,9mg/dl, vitamina B12:247pg/ml, ácidofólico: 6,5 mg/dl, resonancia magnética nuclear de encéfalo muestra lesión lacunar secuelar en núcleos de la base del lado derecho. EEG normal. La evaluación neuropsicológica describe una edad mental de 10 años a los 17 años, con un cociente intelectual de 56 puntos y trastorno psicótico breve. Ecocardiograma: regurgitación tricúspide y pulmonar leve sin repercusión hemodinámica.
Recibió tratamiento con risperidona y piridoxinacon mejoría en el cuadro psicótico, aunque persiste con inquietud psicomotora y crisis de ansiedad. Presentó repigmentación discreta de la piel, cabello, pestañas y cejas. Recibió terapia física regular.
DISCUSIÓN
Los mecanismos fisiopatológicos responsables de la homocistinuria, no se conocen con exactitud; sin embargo, la elevación de homocisteína en plasma sería el factor responsable de las principales manifestaciones clínicas multisistémicas, de inicio lento y curso progresivo, esto se debería a la inducción de estrés oxidativo y una incorporación incrementada de homocisteina en los distintos sistemas del organismo (7,8).
El paciente presentó hipopigmentación en piel y faneras, livedo reticularis y rash malar, descritas en los casos de homocistinuria clásica, en la cual el exceso de homocisteína ejercería una acción inhibidora en las enzimas histidasa y tirosinasa interfiriendo en la melanogénesis normal. Se evidenció un oscurecimiento parcial de la piel y faneras luego del tercer mes de tratamiento con piridoxina, que se explicaría por el incremento de la remetilación de la homocisteína a metionina(9).
La subluxación bilateral del cristalino (ectopialentis),asociada a miopía severa y glaucoma, fue diagnosticada y corregida quirúrgicamente a los 6 años; hallazgo oftalmológico presente en más del 80% de casos de homocistinuria clásica(10).La luxación del cristalino, preferentemente de posición inferonasal en homocistinuria, se generaría debido a cambios degenerativos en las fibras zonulares secundarias a la deficiencia de CBS (11). En la homocistinuria, el glaucoma puede presentarse hasta en 23% de casos, lo cual contrasta con otros síndromes a considerar en el diagnóstico diferencial como el síndrome de Weill-Marchesani y el síndrome de Marfan que asocian glaucoma en 80%y 8% respectivamente (12).
El fenotipo marfanoide, incluyendo la dolicoestenomelia, aracnodactilia y la escoliosis; son manifestaciones a nivel de tejido conectivo y esquelético generadas tanto por la deficiencia de cisteína como por el incremento de homocisteína que ocasionan un mal plegamiento de fibrilina tipo I debido a la reducción de enlaces disulfuro en los dominios cbEGF de esta proteína (13,14).La limitación articular, característica en homocistinuria, es un hallazgo clínico que permite diferenciarlo del síndrome de Marfan donde predomina la hiperlaxitud articular. El paciente presentó, además, una fractura espontánea sugerente de osteopenia. Las fracturas patológicas son otro hallazgo clínico diferencial en homocistinuria (Tabla 1), este fenómeno se produciría por la interferencia de los enlaces cruzados de homocisteína con el colágeno(15).
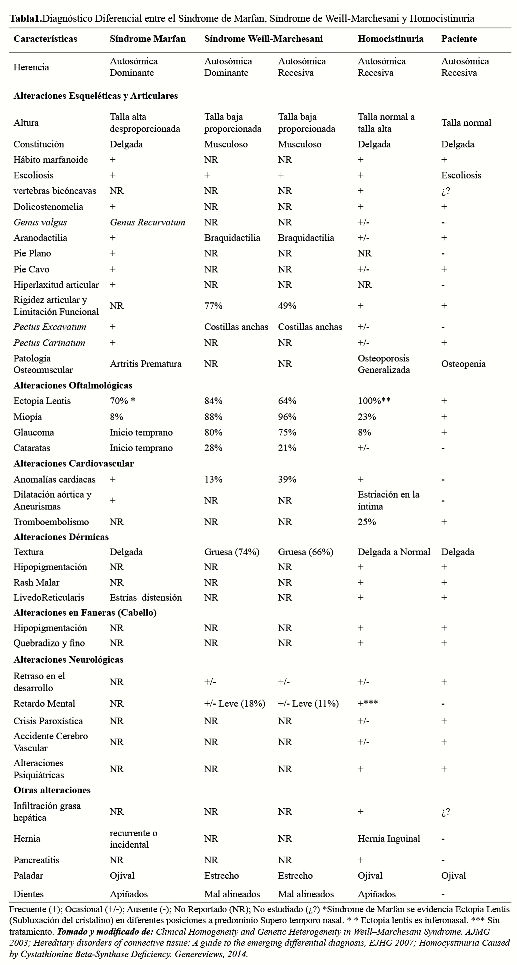
La evaluación neuropsicológica evidenció un retraso mental con coeficiente intelectual de 56 puntos, probablemente debido a la inhibición competitiva del transporte de aminoácidos al cerebro y formación de neurotransmisores, como consecuencia de la elevada concentración de metionina, homocisteína y el déficit de cistationina (16). El trastorno cognitivo en homocistinuria, como el que presenta el paciente, se debería a la actividad neurotóxica con aumento de S- adenosil homocisteína, ácidohomocisteíco y ácido sulfínico de la homocisteína, debido al influjo de calcio promovido por la activación de receptores deglutamatotipo NMDA(17). Aquellos pacientes con homocistinuria que mantienen los valores adecuados de homocisteína(<11 μmol/L) desde temprana edad, antes de los 5 años, se relacionan con coeficientes intelectuales normales (18).El paciente presentó, además, alteraciones de comportamiento y psicosis, que suelen presentarse en la homocistinuria en más del 50% de pacientes adultos, que podrían ser secundario saldaño en la trasulfuración de la homocisteína que lleva a homocistinemia y hipermetioninemia por deficiencia marcada de la actividad decistationina β-sintasa en el sistema nervioso central (19).
La hemiparesia izquierda secuelar en el paciente se debió a un infarto lacunar de capsula interna izquierda, una complicación frecuente en homocistinuria. Al menos 8% de los casos de homocistinuria desarrollaría algún evento tromboembólico cerebrovascular para la edad de 16 años(20). Los desórdenes cerebro vasculares, en especial trombosis arterial y venosa, en los pacientes con homocistinuria estarían en relación al estado de hiperhomocisteinemia, estado protrombótico que incrementa el riesgo de infarto cerebral y venoso. La hiperhomocisteinemia produce disfunción endotelial que disminuye la biodisponibilidad de óxido nítrico derivado del endotelio, afectando especialmente vasos pequeños como los vasos cerebrales(21).
La homocistinuria, en la mayoría de casos, suele pasar desapercibida durante edades muy tempranas; en algunas ocasiones existen signos inespecíficos como falla de medro, retraso psicomotor que deberían ser signos de alarma o sospecha para descarte de esta y otras enfermedades metabólicas. Nuestro paciente presentó estos signos, además de la hipopigmentación de piel y faneras y la limitación articular en extremidades, que no permitieron sospechar en esta enfermedad oportunamente. Estrategias como el tamizaje neonatal(22),aún no disponible en la mayoría de centros hospitalarios en el país, permitirá un diagnóstico y tratamiento oportuno de la homocistinuria y otras enfermedades metabólicas tratables.
REFERENCIAS BIBLIOGRÁFICAS
1.Refsum H, Fredriksen A, Meyer K, Ueland PM, Kase BF. Birth prevalence of homocystinuria. J Pediatr. 2004; 144(6): 830-2. [ Links ]
2. Blom HJ, Smulders Y. Overview of homocysteine and folate metabolism: With special references to cardiovascular disease and neural tube defects. J Inherit Metab Dis. 2011; 34(1):75-81. [ Links ]
3. Picker JD, LevyHL. Homocystinuria caused by cystathionine beta-synthase deficiency. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al(editors). Gene Reviews. Seattle: University of Washington; 1993. [ Links ]
4. Mudd SH, FinkelsteinJD, Irreverre F, LasterL. Homocystinuria: An enzymatic defect. Science. 1964;143(3613): 1443-5. [ Links ]
5.Colorado UO. CBS Mutation Database. Colorado, USA: University of Colorado, Health Science Center;2015. [ Links ]
6. Iacobazzi V,Infantino V, Castegna A, Andria G. Hyperhomocysteinemia: related genetic diseases and congenital defects, abnormal DNA methylation and newborn screening issues. Mol Genet Metab. 2014;113(1-2):27-33. [ Links ]
7. Jakubowski H. Molecular basis of homocysteine toxicity inhumans. CMLS. 2004;61(4): 470-87. [ Links ]
8. Ungvari Z, Csiszar A, Edwards JG, Kaminski PM, Wolin MS, Kaley G, et al. Increased superoxideproduction in coronary arteries in hyperhomocysteinemia: role of tumor necrosis factor-alpha, NAD(P)H oxidase, and inducible nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2003;23(3):418-24. [ Links ]
9. Reish O, Townsend D, Berry SA, Tsai MY, King RA. Tyrosinase inhibition due to interaction of homocyst(e)ine with copper: the mechanism for reversible hypopigmentation inhomocystinuria due to cystathioninebeta-synthase deficiency. Am J Hum Genet. 1995; 57(1): 127-32. [ Links ]
10. Gerding H. Ocular complications and a new surgical approach to lens dislocation in homocystinuriadue to cystathionine-beta-synthetase deficiency. Eur J Pediatr. 1998;157 (S2):S94-101. [ Links ]
11. Chandra A,Charteris D. Molecular pathogenesis and management strategies of ectopialentis. Eye. 2014; 28(2):162-8. [ Links ]
12. Sadiq MA,Vanderveen D. Genetics of ectopialentis. Seminars in ophthalmology. 2013;28(5-6):313-20. [ Links ]
13. Whiteman P, Hutchinson S, Handford PA. Fibrillin-1 misfolding and disease. Antioxid Redox Signal. 2006; 8(3-4): 338-46. [ Links ]
14. Hutchinson S, Aplin RT, Webb H, Kettle S, Timmermans J, Boers GH, et al. Molecular effects of homocysteine on cbEGF domain structure: insights into the pathogenesis of homocystinuria. Journal of molecular biology. 2005; 346(3): 833-44. [ Links ]
15.Castejon MG, Pallardo Y. Manifestaciones esqueléticas en la homocistinuria. Rev Esp Cir Osteoart. 1994; 29: 321-6. [ Links ]
16. Yap S, Rushe H, Howard PM, Naughten ER. The intellectual abilities of early-treated individuals with pyridoxine-nonresponsive homocystinuria due to cystathionine beta-synthasedeficiency. J Inherit Metab Dis. 2001;24(4):437-47. [ Links ]
17. Arzumanyan ES. Mechanisms of homocyestic acid neurotoxicity. Neurochem J. 2010; 4(3):222-7. [ Links ]
18. Biancheri R, Cerone R, Rossi A, Schiaffino MC, Caruso U, Minniti G, et al. Early-onsetcobalamin C/D deficiency: epilepsy and electroencephalographic features. Epilepsia. 2002; 43(6):616-22. [ Links ]
19. Abbott MH, Folstein SE, Abbey H, Pyeritz RE. Psychiatric manifestations of homocystinuriadue to cystathioninebeta-synthase deficiency: prevalence, natural history, and relationship to neurologic impairment and vitaminB6-responsiveness. Am J Med Genet. 1987; 26(4):959-69. [ Links ]
20. Skovby F, Gaustadnes M, Mudd SH. A revisit to the natural history of homocystinuriadue to cystathionine beta-synthase deficiency. Mol Genet Metab.2010; 99(1):1-3. [ Links ]
21. Lentz SR.Mechanisms of homocysteine-induced atherothrombosis. J Thromb Haemost.2005;3(8):1646-54. [ Links ]
22. García M, Baldellou A. Homocystinuria,a great stranger? Keys for the primary carediagnosis. Acta Pediatr Esp.2009; 67(11):535-41. [ Links ]
Agradecimientos
NIH Research Training Grant #R25 TW009345, por apoyo alentrenamiento de investigadores.
Fuentes de financiamiento
Presupuesto de Investigación del InstitutoNacional de Ciencias Neurológicas.
Declaración de conflictos de interés
Los autores declaran no tener conflictos deinterés en la publicaciónde este artículo.
Correspondencia
Mario Cornejo-Olivas
Centro de Investigación Básica enNeurogenética
Instituto Nacional de Ciencias Neurológicas
Jr. Ancash 1271, Barrios Altos, Lima 1, Perú.
Teléfono:+51 1 328 51 89.
Correo electrónico: mario.cornejo.o@incngen.org.pe
Recibido:22/09/2015
Aceptado:23/11/2015
