










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Neuro-Psiquiatría
versión impresa ISSN 0034-8597
Rev Neuropsiquiatr vol.78 no.4 Lima oct. 2015
MELAS en el Perú: reporte de caso.
MELAS in Peru: case report.
Carlos Escalante-Gavancho1,a, Jorge A. Vattuone-Echevarría2,b,Elizabeth Candia-Rivera2,b, Jaime Isidro- Huarcaya1, a, Jorge Escalante-Canorio1,a.
Departamento de Neuropediatria, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
2 Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
a Médico Neuropediatra ; b Médico Residente de Neurología.
RESUMEN
El síndrome de MELAS es una rara citopatía mitocondrial de difícil diagnóstico. Reportamos el caso de una niña de 10 años, que ingresó al Instituto Nacional de Ciencias Neurológicas deLima, Perú, quien presentó episodios bruscos similares a accidentes cerebrovasculares y crisis epilépticas. Los estudios de neuroimágenes mostraron infartos y el examen genético fue positivo para MELAS identificando la mutación más frecuenteA3243G.
PALABRAS CLAVE: Infarto, mutación A3243G, acidosis láctica, miopatíamitocondrial.
SUMMARY
MELAS syndrome is a rare mitochondrial cytopathy difficult to diagnose. We report the case of a 10year old girl who was admitted to the National Institute of Neurological Sciences of Lima - Peru, who presented sudden stroke like episodes and seizures. Neuroimaging studies showed infarction and genetic testing was positive for identifying the most common MELAS mutation(A3243).
KEYWORDS: Infarction, mutation A3243G,lactic acidosis,mitochondrial myopathy.
INTRODUCCIÓN
La encéfalomiopatía mitocondrial, con acidosis láctica y episodios tipo ictus (MELAS, Mitochondrial encephalomyopathy, lacticacidosis, and stroke-likeepisodes)es una entidad clínica rara de herencia mitocondrial que fue descrita por primera vez por Pavlakis et al., en 1984 (1). El cuadro clínico muestra déficits neurológicos repentinos que son llamados episodios de tipo ictus recurrente (hemiparesia, hemianopsia y / o ceguera cortical), crisis epilépticas y cefalea recurrente. Además puede haber nauseas, vómitos, diabetes mellitus, baja estatura, debilidad muscular, pérdida auditiva neurosensorial y acidosisláctica(2). La miocardiopatía se puede desarrollar en las últimas etapas de la enfermedad (3,4).
La enfermedad MELAS es causada por mutaciones del genoma mitocondrial, en el genMT-TL1 que codifica ARNt Leu, siendo la mutación más común A3243G hasta en un 80% (3,5). Otras mutaciones también están asociadas (G3244A, T3258C, C3256T,T3271C, T3291C). MELAS está dentro de las enfermedades mitocondriales que ocurren con mayor frecuencia. La bioquímica de los defectos mitocondriales puede mostrar acidosis láctica y en la biopsia muscular se puede observar Fibras Rojas Rasgadas (RRF), NADH yDHS. La nicotinamina-adenina-dinucleótidotretazolium-reductasa (NADH) y la dehidrogenasa succínica (DHS) son enzimas oxidativas mitocondriales. LaDHS tiñe tanto mitocondrias normales como anormales, mientras que NADH sólo tiñe las mitocondrias normales, incremento de la cantidad de lípidos intermiofibrilares, y alteraciones morfológicas y de tamaño de las mitocondrias (6).
En las neuroimágenes, la TAC cerebral puede mostrar calcificaciones bilaterales en los núcleos de la base. En la RM de encéfalo inicial o evolutiva los hallazgos son lesiones hiperintensas en imágenes potenciadas en T2,localizadas predominantemente en la región cortical de los lóbulos occipitales, parietales y temporales, que no corresponden claramente a un único territorio vascular, asimismo se puede observar atrofiacerebelosa y en la RM con espectroscopia, se reporta disminución de la N-acetil aspartato y aumento de lactato (7).
Actualmente no hay tratamiento curativo, sin embargo se ha reportado que la suplementación con antioxidantes, sustratatos y cofactores de la cadena respiratoria en forma de vitaminas como la tiamina(B1), rivoflavina (B2),nicotinamida (niacinamida), coenzima Q10 (ubidecarenona),idebenona, succinato,citocromo C, menadiona (vitaminaK3), ácido ascórbico, (vitaminaC), ácido tioctico (ácido alfa lipoico),corticoides, coenzima Q puede ser clínica y bioquímicamente beneficiosa (8).
Caso clínico
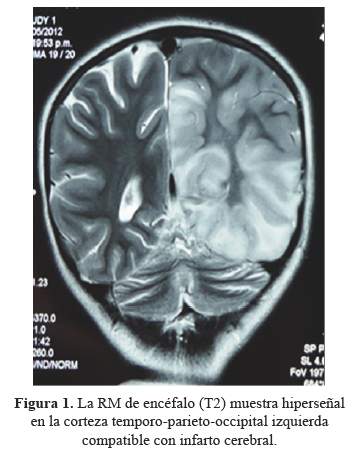
Niña de 10 años de edad sin antecedentes patológicos de importancia, que presenta de manera súbita estado confusional, cefalea holocraneana asociada a vómitos explosivos, sensación de alza térmica y crisis tónico clónico generalizada. Ingresa a emergencia del Instituto Nacional de Ciencias Neurológicas (INCN), con cuadro de un día de evolución de inicio súbito y curso progresivo caracterizado por desorientación, dificultad para la marcha, disminución de fuerza de hemicuerpo izquierdo, cefalea intensa asociada a vómitos explosivos, alza térmica, dolor abdominal, alucinaciones visuales, crisis tónico clónico generalizada y crisis parciales motoras en hemicuerpo izquierdo en dos oportunidades. En la Resonancia Magnética Cerebral (RM),corte axial y coronal en T2 y FLAIR se observan los signos de infarto cerebral izquierdo (Figura 1).
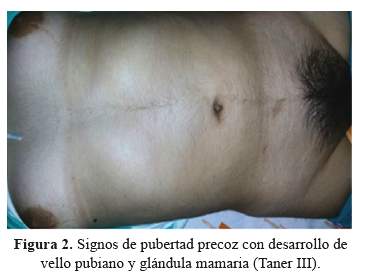
En la exploración general se observó regular estado general, buen estado de nutrición e hidratación, signos de pubertad precoz caracterizados por hipertricosis, vello pubiano y axilar (TanerIII), mamas en TanerIII (Figura 2), talla baja para la edad (127,5 cm) la cual está por debajo del percentil 3 para la edad). En el examen neurológico despierta, desorientada, no obedecía órdenes, movilizaba cuatro extremidades con fuerza normal, tono conservado, reflejos osteotendinosos2/4, hiperestesia generalizada, Babinski bilateral, hipoacusia bilateral, rigidez de nuca ++, noKernig, ni Bruzinski.
Los exámenes auxiliares mostraron los siguientes resultados, el estudio citoquimico en LCR fue normal, el lactato de 6,64 mmol/L (intervalo de referencia 1,13-3,23 mmol/L) y el lactato en suero 4,62 mmol/L (valor de referencia es inferior a 2,2 mmol/l);potenciales evocados auditivos ausentes, potenciales visuales normales y la evaluación neuropsicológica concluyó en CIT de 50 correspondiente a categoría intelectual deficiente con una edad mental de 4 años y10 meses.
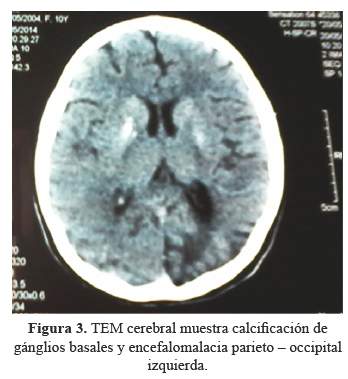
La TEM cerebral en corte axial se evidencia calcificaciones de aspecto irregular en ganglios basales bilateral, imagen hipodensa no homogénea occipito-parietal izquierdo, condiciona retracción del asta ventricular adyacente. Resto de estructuras parenquimales de hemisferios cerebrales, diencéfalo, cerebelo y tronco de caracteres normales (Figura3).
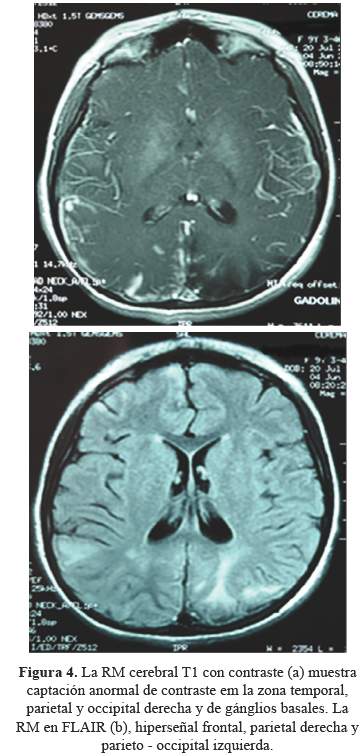
La resonancia magnética cerebral muestra en T1con contraste y FLAIR, captación anormal de contraste y señala normal a nivel cortical y subcortical frontal, temporal, parietal y occipital derecho, además de ambos ganglios basales así como encefalomalacia parietal y occipital izquierda (Figura 4).
Espectroscopia cerebral por resonancia magnética de caracteres normales, específicamente no se observó presencia de lactato. La angio resonancia de cerebro fue de caracteres normales.
Se realizó un estudio genético a partir del DNA, mediante técnica de PCR-RLFP y posterior tinción con bromuro de etilo, encontrando una mutación puntual en el DNA mitocondrial de la paciente y de su madre en A3243G con un porcentaje de heteroplasmia de alrededor del 50% para la hija y del 5% a 10% para la madre.
La paciente evolucionó favorablemente, ha presentado episodios de cefaleas que ceden con analgésicos comunes y se desenvuelve en forma independiente. Recibe tratamiento con carbamazepina, coenzima Q 10, vitamina E 400 UI y triptorelina.
DISCUSIÓN
MELAS es un desorden mitocondrial de presentación heterogénea causada por defectos en la producción de energía intracelular, que afecta a los tejidos con alto requerimiento energético (músculo y cerebro). El diagnóstico requiere estudios de biología molecular, genética, imágenes y clínica.
Desde el punto de vista epidemiológico en Inglaterra se describe una incidencia global de enfermedades mitocondriales en un 12,48 por 100 000habitantes (9). La prevalencia absoluta de la mutación A3243G ha sido estimada en 0,06% por cada 100 000 individuos en la población general(9,10). En un estudio Finlandés en niños la frecuencia fue de18,4 por cada 100 000 individuos (10) y en adultos de 16,3/100000 (11). En Japón la prevalencia fue de 0,18 /100 000 de la población total, de 0,5/100 000 en menores de 18 años y de0,12/100 000 en la población mayor de 18 años (5). El presente es el primer caso publicado en el Perú.
El caso clínico que presentamos, tiene confirmado el diagnóstico de MELAS en base al estúdio clínico, radiológico y genético que mostró una mutación puntual en A3243G, que de acuerdo a la bibliografía es la mutación más frecuente, hasta en un 80% (3,4). En la madre también se encontró la mutación descrita en un 5 a 10%, lo que explica la ausencia de manifestaciones clínicas por el bajo porcentaje de mutación, a diferencia de la paciente que tiene la mutación en alrededor del 50%. MELAS se presenta en un 79% antes de los20 años de edad, más frecuentemente entre los dos y diez años (12), es caracterizada por una clínica variada con frecuente afectación multisistémica. Caracterizada por talla baja, epilepsia focal o generalizada y episodios de stroke like con afectación frecuente occipitotemporoparietal, acidosis láctica sérica y en líquido cefalorraquídeo. Otras manifestaciones son diabetes, sordera, deterioro cognitivo, ataxia, miopatía, retinopatía pigmentaria, cefalea recurrente (migraña like), entre otros. En relación al caso presentado esta paciente debutó con súbito trastorno de sensorio, cefalea asociada a vómitos explosivos, sensación de alza térmica, crisis tónico clónico generalizada y hemiparesia izquierda. Además presentó signos de pubertad precoz, que no es característico de MELAS, talla baja, hipoacusia bilateral y manifestaciones de un episodio de infarto cerebral caracterizado por: hemiparesia izquierda, hiperestesia generalizada, Babinski bilateral y hemianopsia homónima derecha como se describe en esta citopatíamitocondrial (2). En cuanto a las manifestaciones clínicas endocrinológicas relacionadas a MELAS por mutación que afecta al RNAt se describe especialmente diabetes miellitus, hipertiroidismo y talla baja, el caso presentado se vio asociado a pubertad precoz hecho que lo hace aún más particular, a pesar que en realidad se podrían presentar otras manifestaciones que afecten la regulación de cualquiera de los ejes del sistema endocrino.
Los hallazgos neuroimagenológicos en la tomografía cerebral son inespecíficos pudiendo visualizarse calcificaciones o infartos de núcleos basales. En este caso se observa calcificación bilateral de ganglios basales y una lesión encefalomalacica en la región parietooocpital izquierda. La RMN evidencia lesiones hiperintensas en imágenes potenciadas en T2 que afectan fundamentalmente a la corteza de lóbulos occipitales, parietales y temporales, que también se observan en la paciente (7).
En cuanto al diagnóstico genético, la disfunción mitocondrial en el MELAS se origina por mutación del gen MT-TL1 del ADN mitocondrial, en la mayor parte de los casos la mutación es en la transición adenina guanina en la posición 3243 (A3243G) en la región que codifica para el RNAt Leu. El RNAt mitocondrial es el factor de transmisión de la transcripción mitocondrial. Esta mutación repercutiría en las sub unidades con mayor contenido de leucina como el complejo de la cadena respiratoria (MT ND3 yMT-ND6) con consecuente disminución en el gradiente electroquímico a través de la membrana mitocondrial interna y disminución de la síntesis de ATP. Otra hipótesis es que el modelo mutado de RNAt Leu. (UUR) conllevaría a la pérdida de la especificidad de la leucil-transferasa para la sustitución de la leucina por fenilalanina o una terminación prematura de la traducción. Se sabe que la mutación más comúnes la A3243G hasta en un 80%(3,5). En el análisis genético del DNA mitocondrial de esta paciente se encuentra un porcentaje de heteroplasmia de alrededor del 50 %.
Respecto a la terapia, en cuanto no se conozca los mecanismos específicos de los efectos de la mutación, únicamente estaría restringido a observar la respuesta clínica favorable en algunos casos. Los esquemas terapéuticos en MELAS se basan enantioxidantes, sustratatos y cofactores de la cadena respiratoria en forma de vitaminas como la tiamina (B1), rivoflavina(B2), nicotinamida (niacinamida),coenzima Q10 (ubidecarenona), idebenona, succinato, citocromo C, menadiona ( vitamina K3),ácido ascórbico,(vitamina C), ácido tioctico (ácidoalfa lipoico), corticoides (8). En el caso de la paciente se usa coenzima Q 10,vitamina E y triptorelina, con evolución estable.
CONCLUSIONES
MELAS es una enfermedad mitocondrial de presentación poco frecuente y sintomatología compleja con afectación multisistémica que puede llevar a error diagnóstico si no se tiene en mente la posibilidad de esta entidad cuyas principales manifestaciones son encefalopáticas y de ictus. El diagnóstico preciso requiere de estudios genéticos además de los de laboratorio y por imágenes. La efectividad de los medicamentos antioxidantes, los que modifican la función de la cadena respiratoria y los que reducen el acúmulo de metabolitos, entre otros, es incierta.
REFERENCIAS BIBLIOGRÁFICAS
1. Pavlakis SG, Phillips PC, Di Mauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol. 1984;16(4):481-8. [ Links ]
2. Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes:basic concepts, clinical phenotype,and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008;1142:133-58. [ Links ]
3. Ito H, Mori K, Kagami S. Neuroimaging of stroke-like episodes in MELAS. Brain Dev. 2011;33(4):283-8. [ Links ]
4. Hsu YC, Yang FC, Perng CL, Tso AC, Wong LJC, Hsu CH. Adult-onset of Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes (MELAS) Syndrome presenting as acute meningoencephalitis: A Case Report. J Emerg Med. 2012;43(3):e163-6. [ Links ]
5. Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T, et al. MELAS: A nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta BBA Gen Subj. 2012;1820(5):619-24. [ Links ]
6. Koo B, Becker LE, Chuang S, Merante F, Robinson BH, MacGregor D, et al. Mitochondrial encephalomyopathy, lacticacidosis, stroke-like episodes (MELAS): clinical, radiological,pathological, and genetic observations. Ann Neurol.1993; 34(1): 25-32. [ Links ]
7.Cano A, Romero AI, Bravo F, Vida JM, Espejo S. Síndrome MELAS: hallazgos neurorradiológicos. Radiología. 2002; 44(2): 69-74. [ Links ]
8. Abe K, Matsuo Y, Kadekawa J, Inoue S, Yanagihara T. Effect of coenzyme Q10 in patients with mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS): Evaluation by noninvasive tissue oximetry. J Neurol Sci.1999; 162(1): 65-8. [ Links ]
9. Chinnery PF, Turnbull DM. Epidemiology and treatment of mitochondrial disorders. Am J Med Genet. 2001; 106(1):94-101. [ Links ]
10. Uusimaa J, Moilanen JS, Vainionpaa L, Tapanainen P, Lindholm P, Nuutinen M, et al. Prevalence, segregation, andphenotype of the mitochondrial DNA 3243A>Gmutation in children. Ann Neurol. 2007; 62(3):278-87. [ Links ]
11. Majamaa K, Moilanen JS, Uimonen S, Remes AM, Salmela PI, Karppa M, et al. Epidemiology of A3243G,the mutation for mitochondrial encephalomyopathy, lacticacidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am J Hum Genet. 1998;63(2):447-54. [ Links ]
12. Di Mauro S, Hirano M. MELAS. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al. (editores). Gene Reviews. Seattle, Washington: University of Washington; 1993. [ Links ]
Fuente de financiamiento: Autofinanciado.
Conflictosde interés: Los autores declaran notener conflictos de interésen la publicación del presenteartículo.
Correspondencia:
Carlos Escalante Gavancho.
Instituto Nacional de Ciencias Neurológica
Jr. Ancash 1271. Lima, Perú
Teléfono:51941366983
Correoelectrónico: drescalantespn@yahoo.es
Recibido:07/09/2015
Aceptado:07/12/2015
