










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Neuro-Psiquiatría
versión impresa ISSN 0034-8597
Rev Neuropsiquiatr vol.79 no.1 Lima ene. 2016
REPORTE DE CASO
Síndrome de Aicardi: Presentación de un caso clínico y revisión de la literatura.
Aicardi’s syndrome: A case report and literature review.
Miguel A. Villarreal-Ybazeta1,a, Felicitas A. Tirado-Chavarría1,a, Nila E. Calderón-Alarcón2,b
1 Hospital Regional Docente de Trujillo. La Libertad, Perú.
2 Instituto Regional de Oftalmología. La Libertad, Perú.
a Médico Pediatra ; b Médico Oftalmólogo Pediatra.
RESUMEN
El síndrome de Aicardi (SA), es una rara enfermedad neurogenética, diagnosticada clínicamente por la triada clásica de agenesia del cuerpo calloso, espasmos infantiles y lagunas corioretinales. El objetivo de nuestro trabajo fue presentar el espectro clínico y la evolución de este trastorno en un caso. Reportamos una lactante de dos meses, de malformaciones cerebrales múltiples, que ingresa a emergencia por presentar crisis convulsivas. Durante la hospitalización, los hallazgos clínicos – electroencefalográficos, neuroimágenes y evaluación oftalmológica confirman el diagnóstico de SA. Se resalta la importancia de conocer las manifestaciones clínicas del síndrome, para tenerlo en cuenta como diagnóstico diferencial, sobre todo en aquellos casos que presenten espasmos infantiles asociados a agenesia del cuerpo calloso.
PALABRAS CLAVE: agenesia del cuerpo calloso, espasmos infantiles, lagunas coriorretinales, síndrome de Aicardi.
SUMMARY
Aicardi’s syndrome (SA) is a rare genetic disease, clinically characterized by the classic triad of agenesis of the corpus callosum, infantile spasms and chorioretinal lacunae. The aim of this presentation is to describe the clinical spectrum and evolution of the disorder. We report the case of a 2-month old female infant with a prenatal diagnosis of multiple brain malformations, brought into the Emergency Room while experiencing seizure spells. During her hospitalization, electroencephalography, neuroimaging and ophthalmologic evaluations confirmed the diagnosis of SA. The importance of knowing the clinical manifestations of the syndrome is emphasized, in order to keep it in mind as part of the differential diagnosis for cases presenting infantile spasms associated with agenesis of the corpus callosum.
KEYWORDS: Agenesis of the corpus callosum, infantile spasms, chorioretinal lacunae, Aicardi syndrome.
INTRODUCCIÓN
El síndrome de Aicardi (SA), fue descrito por el profesor Jean Aicardi (1926-2015), hace más de 50 años, basado en el análisis de la observación de pacientes con "espasmos infantiles en flexión", en los cuales se demostró agenesia del cuerpo calloso por neumoencefalografía y anormalidades oculares (microftalmia, coroiditis atrófica y coloboma del nervio óptico) como tercer componente sintomático (1). Unos años más tarde, se hizo una descripción detallada del síndrome, postulando como criterios diagnósticos la triada clásica de espasmos infantiles, agenesia del cuerpo calloso y lagunas coriorretinales (2).
Es una enfermedad rara (OMIM 304050), con herencia dominante ligada al cromosoma X, que afecta exclusivamente a mujeres o varones con síndrome de Klinefelter (XXY), siendo letal en hemicigotos varones (3).
Se desconoce su epidemiología exacta, aunque algunos países como Suiza, Holanda y Estados Unidos han estimado su incidencia en 1/99 000, 1/93 000 y 1/105 000 recién nacidos vivos respectivamente, así como una prevalencia mundial estimada de más de 4 000 pacientes, 10 veces más alta que en reportes anteriores (4).
El objetivo de nuestro trabajo fue describir un caso registrado en nuestra población, y mostrar las clásicas anormalidades oculares, espasmos en flexión y agenesia del cuerpo calloso, como las alteraciones electroencefalográficas, hallazgos de neuroimágenes y evolución clínica del síndrome de Aicardi.
Caso clínico
Lactante, sexo femenino, dos meses de edad, ingresa a emergencia, por ocurrencia de crisis convulsivas consistentes en movimientos involuntarios aparentemente generalizados, con lateralización de la mirada a la derecha, y cianosis perioral de aproximadamente un minuto de duración, luego de la cual se muestra somnolienta y flácida.
Producto de primera gestación, controlada, parto por cesárea, indicada por diagnóstico prenatal de "hidrocefalia fetal", Apgar 9/9, peso al nacimiento de 3400 gramos. No requirió maniobras de reanimación. No se reporta consanguinidad, ni endogamia, estudios para infección materna por el virus de la inmunodeficiencia humana y sífilis no reactivos. Alta conjunta a las 48 horas de vida.
Funciones vitales estables. Cráneo normocéfalo, circunferencia cefálica 37.5 cm, facies no característica. Somnolencia, cuadriflexa, movimientos espontáneos escasos. Reflejos primitivos débiles, reflejos osteotendinosos 2/4, simétricos, hipotonía axial, hipertonía apendicular. Respuesta visual y auditiva aparentemente conservada.
Se hospitaliza para manejo y estudio. Se realizan exámenes hematológicos, bioquímicos, gases arteriales, serología para infecciones perinatales, ecografía abdominal y radiografía de tórax con resultados dentro de límites normales. Electroencefalograma: Intercrítico, en sueño fisiológico, muestra actividad theta, con ondas agudas hipervoltadas, asincrónicas, a cmpredominio de hemisferio derecho, que se alternan con intervalos de supresión de la actividad de base. Resonancia magnética de encéfalo: Agenesia de cuerpo calloso, múltiples quistes inter hemisféricos, quistes de plexos coroideos, polimicrogiria y heterotopias nodulares subependimarias. Estudio oftalmológico de fondo de ojo: Muestra múltiples lesiones coriorretinales lacunares bilaterales. Atrofia sectorial peri papilar del epitelio pigmentario retinal. Coloboma de nervio óptico izquierdo.
Durante la hospitalización, se evidenciaron espasmos infantiles en flexión, que se correlacionaron con el trazado "brote-supresión" del electroencefalograma, considerando síndrome de West sintomático como diagnóstico complementario. El tratamiento anticonvulsivante se inició con ácido valproico, incrementando progresivamente la dosis hasta 70 mg/kg/día, y como segundo fármaco levetiracetam hasta alcanzar una dosis máxima de 60 mg/kg/día, sin conseguir mejoría. Después de la segunda semana de tratamiento, se consideró adicionar vigabatrina en pauta creciente hasta 75 mg/kg/día, con lo que se pudo conseguir una reducción significatica de las crisis, y continuar su tratamiento ambulatoriamente.
DISCUSIÓN
La constelación de síntomas actualmente denominada como síndrome de Aicardi, fue publicada en 1965 por Aicardi y colaboradores, quienes dieron a conocer brevemente, el resultado de la observación de ocho pacientes, con espasmos en flexión, y su asociación con agenesia del cuerpo calloso y anormalidades oculares. En 1969, una nueva publicación de quince pacientes con hallazgos clínicos similares, ampliaron las observaciones originales, reafirmando el descubrimiento de un nuevo síndrome. Señalaron además, que todos sus pacientes eran de sexo femenino, la mayoría de las crisis correspondían a espasmos infantiles, aunque también fueron comunes otros tipos de convulsiones, y el patrón electroencefalográfico predominante correspondía a hipsarritmia atípica. Se documentaron en todos los pacientes, alteraciones neurológicas, como retraso en el desarrollo psicomotor, microcefalia, hipotonía axial, signos piramidales bilaterales, y dificultades deglutorias, que contribuyeron a su mal pronóstico. Aunque se asociaron numerosas anormalidades oculares inespecíficas, las lesiones retinales en sacabocado, denominadas "lagunas coriorretinales", fueron consideradas patognomónicas de esta condición. También se encontraron anomalías esqueléticas y dismorfismos faciales. Además, el síndrome no tenía ninguna etiología genética, infecciosa, o metabólica evidente (1,2).
A pesar de su origen genético, todos los casos conocidos del SA han sido esporádicos, a excepción de un único caso familiar reportado en dos hermanas y otro en dos gemelas monocigóticas. Se piensa que podría estar determinado por una mutación hemicigota "de novo" en el cromosoma X, causando la enfermedad sólo en las mujeres y varones XXY, siendo letal en los varones XY. Aunque la región o gen del cromosoma X no se han identificado plenamente, se lo ha relacionado al locus p22.3, sin que esto se haya confirmado. Debido a que hasta hoy no se ha identificado un marcador biológico asociado a este síndrome, el diagnóstico continúa basándose en una serie de criterios clínicos, electroencefalográficos y de neuroimágenes característicos (3,5,6).
Las neuroimágenes han sido esenciales en la delineación del síndrome, a pesar de que en el momento de su descripción original, sólo estaba disponible la neumoencefalografía. Con el desarrollo de la ultrasonografía, la TC y, especialmente la RM, ha sido posible un mejor análisis de las anomalías. La ultrasonografía fetal de rutina en el segundo trimestre, sobre todo después de la segunda mitad del embarazo, puede detectar malformaciones congénitas del sistema nervioso central, y complementariamente la resonancia magnética puede determinarlas con mayor detalle (7). En el caso que presentamos, un estudio ultrasonográfico prenatal realizado en el tercer trimestre de gestación, determinó una serie de malformaciones cerebrales en el feto, que motivó su referencia para atención hospitalaria del parto (Figura 1).
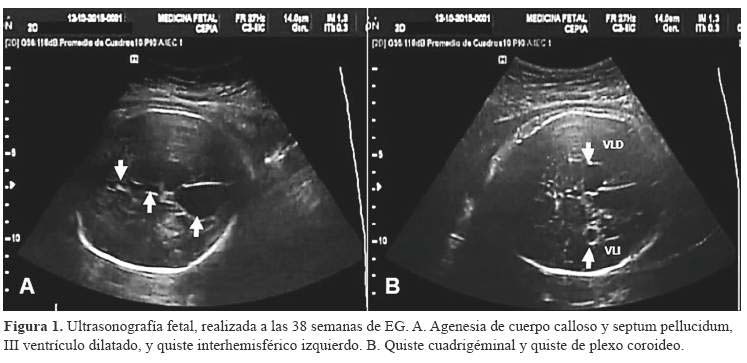
La combinación de estos hallazgos en un feto de sexo femenino, debieron considerar fuertemente al síndrome de Aicardi, que al momento de nacer sería complementada con RM y evaluación oftalmológica para establecer el diagnóstico definitivo (8).
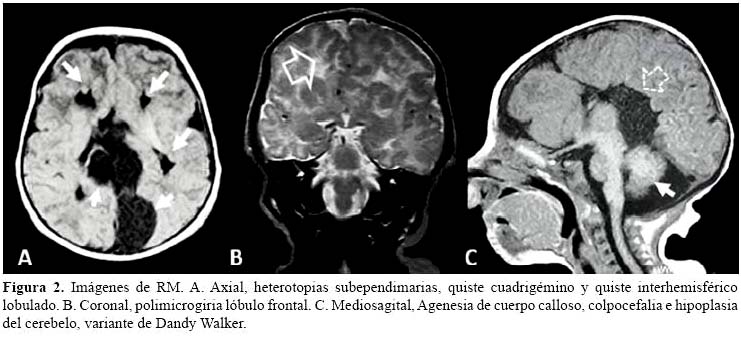
El cuerpo calloso se forma entre la octava y la vigésima semana de gestación, simultáneamente con otras estructuras cerebrales principales. Su agenesia total o parcial, es una malformación que resulta de una agresión que se produce antes del inicio de su desarrollo, y puede presentarse de manera aislada o formar parte de cuadros sindrómicos específicos (9). Con los actuales avances en neuroimágenes, sobretodo la resonancia magnética, cada vez está más claro que la agenesia del cuerpo calloso no es una malformación aislada en el SA, y que las anormalidades asociadas, como ventriculomegalia, asimetría inter hemisférica, heterotopias corticales y peri ventriculares, paquipolimicrogiria, formaciones quísticas (plexos coroideos, tercer ventrículo, región pineal e inter hemisféricas), y papilomas de los plexos coroideos (10), pueden ser tanto o más importantes. En nuestra paciente, la variedad de hallazgos en la resonancia magnética, visualizados prenatalmente por ultrasonografía, permitieron postular al SA, como hipótesis diagnóstica altamente probable (Figura 2).
El hallazgo ocular patognomónico, es un área redondeada hipo pigmentada, conocida como "laguna coriorretinal". Fue el mismo, Aicardi quien describió por primera vez esta lesión, como áreas de "coroiditis atrófica" (pseudotoxoplasmosis), que normalmente se agrupan alrededor del disco óptico. Estas lesiones aparecen de color blanco amarillento, en tamaño variable de medio a tres veces el diámetro del disco, con bordes bien definidos a menudo ligeramente pigmentados, uni o bilaterales, eventualmente inalterables en número o tamaño con el tiempo (2,11). La ausencia de pigmento dentro de las lagunas ayuda a distinguirlas de lesiones similares causadas por coriorretinitis infecciosa (12). Las lagunas coriorretinales han sido raramente observada en otros trastornos, incluyendo el síndrome de banda amniótica y el síndrome oral-facial digital tipo VIII, y en una familia con una forma autosómica dominante rara de microcefalia (13).
Un hallazgo no menos frecuente, es el coloboma del nervio óptico, que puede estar presente en más de la mitad de los casos. Otras anomalías oculares descritas incluyen microftalmia, cataratas, membrana pupilar persistente, coloboma de coroides, sinequias del iris, estafiloma posterior de iris o coroides, anomalías de los vasos retinianos, desprendimiento de retina, hipoplasia del nervio óptico y desarrollo anormal de la fóvea (14). La agudeza visual se puede documentar con pruebas clínicas estándar, tales como las pruebas de Snellen y Sheridan-Gardner, o con el patrón de potenciales evocados visuales (PEV). La afectación de la función visual parece no estar correlacionada con anormalidades del nervio óptico, ni con el número o tamaño de las lagunas coriorretinales, sino más bien con el desarrollo o la extensión de la afectación de la fóvea (15). A pesar de las dificultades propias del paciente, se obtuvieron las imágenes de ambas retinas demostrando las lesiones características y la dimensión del compromiso ocular (Figura 3).
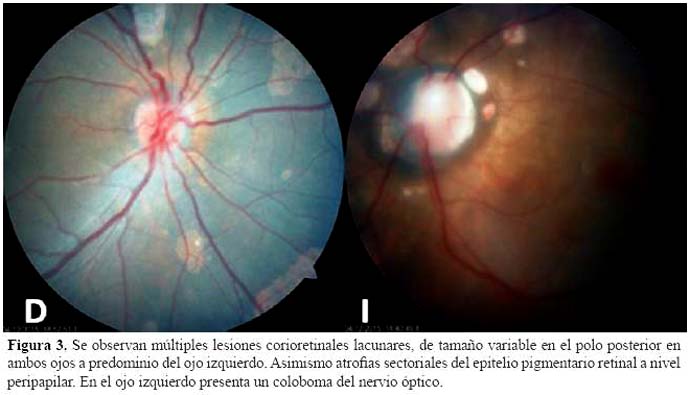
En pacientes con el SA, el patrón clínico de las crisis presenta algunas características que pueden orientar al diagnóstico, entre ellas la aparición precoz de los espasmos infantiles, generalmente antes de la edad de tres meses, los que a menudo son precedidos por crisis clónicas o tónicas focales. Los espasmos suelen ser asimétricos, comprometiendo en mayor medida a un hemicuerpo, fenómeno relativamente infrecuente en casos de otra etiología. Se han descrito alteraciones electroencefalográficas características en este síndrome, particularmente durante la vigilia, el trazado muestra una desorganización de la actividad de base, con un patrón brote-supresión, con completa asincronía entre ambos hemisferios ("split-brain"). Las alteraciones descritas disminuyen y se organizan durante el sueño (16).
Se ha descrito al síndrome de Aicardi, como un trastorno del desarrollo cerebral asociado a la etiología del síndrome de West (17). A nivel nacional, en una serie de encefalopatías epilépticas, descrita por Campos y Cruz, uno de los nueve casos que desarrollaron espasmos infantiles e hipsarritmia, correspondió al síndrome de Aicardi (18). En un estudio más reciente, Guillén y Guillén, reportaron 37 casos con síndrome de West tratados en el sector privado, en Lima, donde 29 de ellos fueron clasificados como sintomáticos, pero ninguno asoció agenesia del cuerpo calloso y lagunas coriorretinales como componentes de la triada clásica del síndrome (19).
La evolución temporal de las crisis acontecidas en nuestra paciente fue característicamente compatible, tanto en la presentación clínica de los espasmos, que se iniciaron a los dos meses de vida, como en la actividad paroxística del EEG (Figura 4).
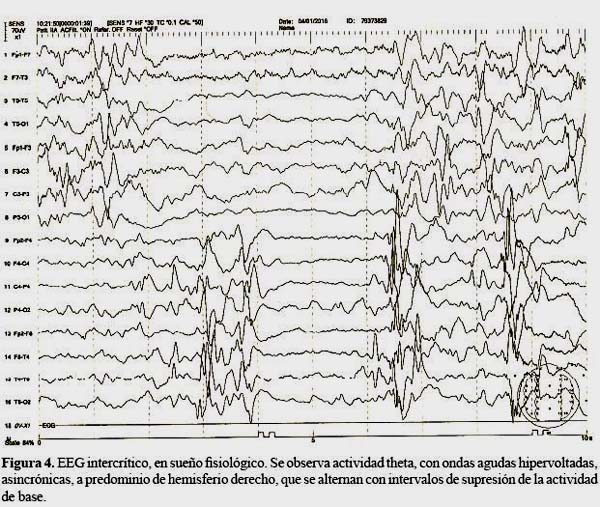
Otras anormalidades extra neurológicas asociadas, como el aspecto facial característico, defectos costovertebrales ("vertebras en mariposa", hemivértebras, costillas faltantes o fusionadas), o tumores (20), no se demostraron en nuestro paciente. A excepción de una lesión vascular cutánea descrita clínicamente como una malformación vascular.
El pronóstico del SA es grave, lo habitual es que curse con epilepsia refractaria y grave discapacidad intelectual. Su mortalidad es elevada en la infancia. Se ha descrito una supervivencia del 76% a los 6 años y del 40% a los 14 años (21). Diferentes complicaciones médicas asociadas a la gravedad de la afectación neurológica, la inmovilidad y la epilepsia refractaria son las principales causas de mortalidad, y se ha descrito como causa principal de ésta las infecciones pulmonares (22, 23).
En conclusión, el conocimiento del espectro clínico del síndrome de Aicardi, a pesar de ser un trastorno poco frecuente, nos permitirá tenerlo en cuenta como diagnóstico diferencial, sobre todo en aquellos casos que presenten espasmos infantiles como manifestación clínica asociada a agenesia del cuerpo calloso, optimizando los recursos diagnósticos y terapéuticos disponibles en nuestro medio.
REFERENCIAS BIBLIOGRÁFICAS
1. Aicardi J, Levebre A, Lerique-Koechlin A. A new syndrome: spasms in flexion, callosal agenesis, ocular anormalities. Electroencephalogr Clin Neurolophysiol. 1965; 19: 609-610. [ Links ]
2. Aicardi J, Chevrie J, Rousselie F. Le syndrome spasmes en flexion, agenesic calleuse, anomalies chorio-retiniennes. Arch Fr Pediat. 1969; 26: 1103-1120. [ Links ]
3. Aicardi J. Aicardi syndrome. Brain Dev. 2005; 27:164-171. [ Links ]
4. Kroner B, Preiss L, Ardini M, Gaillard W. New incidence, prevalence, and survival of aicardi syndrome from 408 cases. J Child Neurol. 2008; 23:531-535. [ Links ]
5. Rosser T. Aicardi syndrome. Arch Neurol. 2003; 60:1471-1473. [ Links ]
6. Fernández J, López E, Simón R, et al. Síndrome de Aicardi: Estudio retrospectivo de una serie de siete casos. Rev Neurol. 2013; 57: 481-488. [ Links ]
7. Bulas D. Fetal magnetic resonance imaging as a complement to fetal ultrasonography. Ultrasound Q. 2007; 23: 3-22. [ Links ]
8. Bromley B, Krishnamoorthy K, Benacerraf B. Aicardi syndrome: prenatal sonographic findings. A report of two cases. Prenat Diagn. 2000; 20: 344-6. [ Links ]
9. Barkovich A, Norman D. Anomalies of the corpus callosum: correlation with further anomalies of the brain. Am J Roentgenol. 1988; 151: 171-179. [ Links ]
10. Hopkins B, Sutton V, Lewis R, Veyver I, Clark G. Neuroimaging aspects of Aicardi syndromes. Am J Med Genet. 2008; 22: 2871-2878. [ Links ]
11. Puertas D, Lozano M, De Domingo B, Ruiz M, González L, Fernández J. Coriorretinopatía lacunar como presentación de síndrome de Aicardi en el lactante. Arch Soc Esp Oftalmol. 2007; 82: 311-314. [ Links ]
12. Shimada Y. Pseudo Toxoplasmosis. In: Djurkovic O. Toxoplasmosis recent advances. Rijeka: In Tech; 2012. p. 173-186. [ Links ]
13. Carney S, Brodsky M, Good W, Glasier C, Cunniff C. Aicardi syndrome: More than meets the eye. Surv Ophthalmol. 1993; 37: 419-424. [ Links ]
14. Fruhman G, Eble T, Gambhir N, Sutton V, Van den Veyver I, Lewis R. Ophthalmologic findings in Aicardi syndrome. JAAPOS. 2012; 16: 238-41. [ Links ]
15. Menezes A, Lewis T, Buncic J. Role of ocular involvement in the prediction of visual development and clinical prognosis in Aicardi syndrome. Br J Ophthalmol. 1996; 80: 805-812. [ Links ]
16. Aicardi J, Chevrie J. Aicardi syndrome. In: Lassonde M, Jeeves M. Callosal Agenesis: A natural split brain, Advances in Behavioral Biology. New York: Plenum Press; 1994. p. 7-17. [ Links ]
17. Campistol J, García A. Síndrome de West. Análisis, factores etiológicos y opciones terapéuticas. Rev Neurol. 2003; 37: 345-352. [ Links ]
18. Campos P, Cruz G. Encefalopatías epilépticas de la infancia. Arq Neuropsiquiatr. 1993; 51:59-65. [ Links ]
19. Guillén-Pinto D, Guillén-Mendoza D. Síndrome de West, experiencia con una serie de casos con acceso al tratamiento de primera línea, en Lima. Rev Neuropsiquiatr. 2015; 78: 65-72. [ Links ]
20. Sutton V, Hopkins B, Eble T, Gambhir N, Lewis R, VanDer-Veyver I. Facial and physical features of aicardi syndrome: Infants to teenagers. Am J Med Genet. 2005; 138: 254-258. [ Links ]
21. Rosser T, Acosta M, Packer R. Aicardi syndrome: spectrum of disease and long-term prognosis in 77 females. Pediatr Neurol. 2002; 27: 343-6. [ Links ]
22. Menezes A, MacGregor D, Buncic J. Aicardi syndrome: Natural history and possible predictors of severity. Pediatr Neurol. 1994; 11: 313-318. [ Links ]
23. Galdós M, Martínez R, Prats J. Síndrome de Aicardi: Variabilidad fenotípica y factores pronósticos. Arch Soc Esp Oftalmol. 2008; 83:29-36. [ Links ]
Recibido: 29/01/2016
Aceptado: 04/03/2016
