










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Neuro-Psiquiatría
versión impresa ISSN 0034-8597
Rev Neuropsiquiatr vol.79 no.3 Lima jul. 2016
ARTÍCULO DE REVISIÓN
Síndrome de Joubert.
Joubert syndrome.
Ana M. Álvarez-Sanz1,2,a, Lizeth Y. Cabanillas-Burgos1,2,b, Ximena P. Huamani-Condori3,c
1 Universidad Nacional de San Agustín. Arequipa, Perú.
2 Hospital Regional Honorio Delgado Espinoza. Arequipa, Perú.
3 Universidad Católica de Santa María. Arequipa, Perú.
a Neuróloga Pediatra ; b Residente de Pediatría ; c Estudiante de Medicina Humana.
RESUMEN
El Síndrome de Joubert (SJ) es un raro trastorno autosómico recesivo con una incidencia de 1/100 000 a 1/150 000 nacidos vivos, considerado una ciliopatía que muestra tubulopatía renal, inmunodeficiencia y trastorno de la migración neuronal en cerebelo y tronco encefálico. El criterio diagnóstico más saltante es el “Signo del Molar” detectado por resonancia magnética cerebral, que identifica hipoplasia de los pedúnculos y vermis cerebeloso. Hasta el momento se han descrito seis subgrupos fenotípicos: SJ puro; SJ con defecto ocular (distrofia retiniana); SJ con defectos renales (no asociados a patología retiniana); SJ con defectos óculorrenales; SJ con defecto hepático y SJ con defectos orofaciodigitales (por ejemplo, lengua bífida, hamartomas múltiples, múltiples frenillos orales y polidactilia). La importancia de un diagnóstico precoz del SJ se puede reflejar en un posible mejor pronóstico y en la posibilidad de mejorar la calidad de vida del paciente con un manejo multidisciplinario y consejo genético para la prevención de nuevos casos en familias afectadas.
PALABRAS CLAVE: Enfermedades del cerebelo, Síndrome de Joubert.
SUMMARY
The Joubert Syndrome (JS) is a rare autosomal recessive disorder with an incidence of 1 / 100000 to 1/150 000 births, considered a ciliopathy and showing renal tubular disease, immunodeficiency and impaired neuronal migration in the cerebellum and brain stem. The main diagnostic criterion is the identification of the “Molar sign” through cerebral magnetic resonance that identifies hypoplasia of cerebellar vermis and peduncles. So far six phenotypic subgroups have been described: JS pure; JS with ocular defect (retinal dystrophy); JS with kidney defects (not associated with retinal pathology); JS with oculorrenal defects; JS with liver defect, and JS with orofaciodigital defects (eg, split tongue, multiple hamartomas, multiple oral frenums and polydactyly). The importance of diagnosing JS at an early stage is related to a possibly better prognosis, and the possibility of improving the patient’s quality of life by means of a multidisciplinary management and provision of genetic counseling for prevention of new cases in affected families.
KEY WORDS: Diseases of the cerebellum, Joubert Syndrome.
INTRODUCCIÓN
Se conoce como Síndrome de Joubert (SJ) (Número OMIM 213300), a la descripción clínica radiológica descrita por Marie Joubert en 1969, que se caracterizaba una malformación cerebelosa, con apnea- hiperpnea episódica neonatal, movimientos oculares anormales, ataxia y retraso mental. Habiéndose notificado hasta el momento poco más de 200 reportes de casos a nivel mundial. Presentamos esta revisión con la finalidad de establecer el diagnóstico a temprana edad, realizar seguimiento clínico para identificar la posible afección de otros órganos y realizar el consejo genético teniendo presente que la enfermedad es transmitida de forma autosómica recesiva con un riesgo de expresión de un 25% cuando hay un hijo afectado.
Generalidades
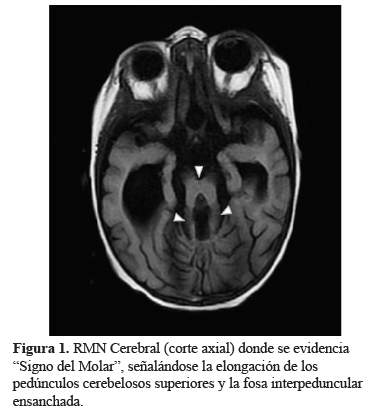
El Síndrome de Joubert fue descrito por primera vez (1), como un trastorno clínico, genético y radiológicamente heterogéneo (2). Se estudiaron dos familias con un fenotipo similar, agenesia del vermis cerebeloso, hiperpnea episódica neonatal, movimientos oculares anormales, ataxia, retraso mental y encefalomeningocele occipital en uno de ellos. En 1977, Boltshauser e Isler describieron esta afección denominándose Síndrome de Joubert - Boltshauser cuyo diagnóstico incluye los hallazgos clínicos con imágenes características en cortes axiales de resonancia magnética, el denominado “signo del molar”(Figura 1) (3), además de la agenesia vermiana (4,5).
Prevalencia y características clínicas
El SJ tiene una prevalencia de 1/100 000 a 1/150 000 nacidos vivos, y se manifiesta desde la etapa neonatal con trastornos en el ritmo respiratorio, nistagmus y alteraciones en la deglución. En el lactante predomina la hipotonía y posteriormente la ataxia cerebelosa. Puede haber apraxia oculomotora, nistagmus, coloboma y epilepsia. Dentro de los rasgos faciales distintivos se encuentran la frente prominente, epicanto, ptosis palpebral y baja implantación de pabellones auriculares.
Genética y fisiopatología
El SJ es una enfermedad de transmisión genética, autosómica recesiva, detectándose hasta hoy 10 genes asociados: AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B y CC2D2A (6). Mutaciones en los genes mencionados codifican proteínas ciliares primarias alteradas, las cuales están asociadas con el funcionamiento de los cilios primarios. Estas organelas juegan un papel fundamental en el funcionamiento de fotorreceptores retinianos y en la señalización implicada en la proliferación celular neuronal de las células del túbulo renal y de los conductos biliares. Diferentes proteínas interaccionan para formar complejos necesarios para la formación de los cilios, que a su vez tienen varias funciones, por ejemplo, en el epitelio renal sirven como sensores celulares (osmolaridad urinaria), intervienen en la proliferación y polaridad celular; una alteración a este nivel conlleva la formación de quistes (7). A nivel encefálico, durante el desarrollo del cerebelo, específicamente del vermis y tallo encefálico, los cilios primarios regulan algunas vías embrionarias implicadas en los principales procesos del desarrollo de proliferación neuroblástica y migración axonal, incluyendo la diferenciación de las células de Purkinje y neuronas granulares (8). Las múltiples funciones de estos cilios en diferentes órganos y tejidos explican por qué diversas mutaciones en estos genes pueden estar asociadas con diversas variantes clínicas.
Clasificación
Se clasifican en seis subgrupos fenotípicos(9):SJ puro; SJ con defecto ocular (distrofia retiniana); SJ con defectos renales (no asociados a patología retiniana); SJ con defectos oculorrenales; SJ con defecto hepático y SJ con defectos orofaciodigitales (por ejemplo, lengua bífida, hamartomas múltiples, múltiples frenillos orales y polidactilia). Puede asociarse también hamartoma hipotalámico y ausencia congénita de glándula pituitaria.
Neuroimagen
Si bien en el SJ existe una gran variabilidad en la presentación clínica, su hallazgo fundamental en la resonancia cerebral es la presencia de distintos grados de displasia cerebelosa con una característica en común, el “Signo del Molar” (“SM”) del mesencéfalo. En algunos casos puede presentarse de forma extensa y difusa, con hemisferios cerebelosos y vermis rudimentarios, localizados dentro de una fosa posterior pequeña, con el IV ventrículo en comunicación con una gran cisterna magna, y con el tronco encefálico hipoplásico y el cuerpo calloso agenésico. En otros casos la displasia es más pronunciada y puede acompañarse de diferentes grados de hipoplasia de los hemisferios cerebelosos, así como de aumento de tamaño del IV ventrículo. Por último, la hipoplasia cerebelosa puede ser asimétrica, asociada a la afectación preferente de un hemisferio cerebeloso y a encefalocele parietooccipital. Puede, también, tratarse de una aplasia total de vermis cerebeloso asociada a profundas alteraciones de la unión cervicomedular, incluyendo ausencia completa de la decusación piramidal, displasia de los núcleos olivar y paraolivar, e hipoplasia del núcleo gracilis y del fascículo solitario (10).
Diagnóstico genético y pronóstico
Hasta la fecha, las pruebas genéticas de diagnóstico están disponibles sólo para unos pocos genes, mientras que algunos laboratorios ofrecen pruebas moleculares de los genes conocidos en una base de investigación. En base a los hallazgos observados en la ecografía obstétrica complementada eventualmente con la resonancia magnética fetal, resulta hoy posible el diagnóstico prenatal de SJ durante el tercer trimestre de la gestación (11). El pronóstico del SJ queda condicionado a la gravedad del cuadro respiratorio, en algunos casos las crisis de apnea remiten espontáneamente durante el primer año de vida, y otras requieren asistencia mecánica. Este dato es crucial por la extrema sensibilidad de éstos pacientes a los efectos depresores respiratorios de algunos agentes anestésicos. También las disfunciones hepáticas y/o renales suelen condicionar el pronóstico vital.
CONCLUSIONES
La importancia de diagnosticar el SJ de forma precoz está relacionada con su pronóstico, la posibilidad de mejorar la calidad de vida del paciente con el manejo multidisciplinario y ofrecer un consejo genético para la prevención en familias con un caso previo.
REFERENCIAS BIBLIOGRÁFICAS
1. Joubert M, Eisenring JJ, Robb JP, et al. Familial agenesis of the cerebellar vermis: A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969; 19: 813-25. [ Links ]
2. Parisi M, Glass I. Joubert syndrome and related disorders. In: Pagon R, Adam M, Bird T, Dolan C,Fong C, Sthephens K (editors). Gene Reviews. Seattle:University of Washington; 2003. p. 1993-2013. [ Links ]
3. Bolsthauser E, Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, mental retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977; 8: 57-66. [ Links ]
4. Burguete A, Cabada T, Bacaicoa M, et al. Síndrome de Joubert: hallazgos en resonancia magnética convencional y tensor de difusión. Radiología. 2012;54(3): 279-282. [ Links ]
5. Satran D, Pierpont MEM, Dobyns WB. Cerebellooculo-renal syndromes including Arima, Senior-Loken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet. 1999; 86: 459-69. [ Links ]
6. Valente E, Dallapiccola B, Bertini E. Joubert syndrome and related disorders: 1879-1888. In: Dulac O, Lassonde M, Sarnat H. Handbook of clinical neurology: Pediatric Neurology Part III. Paris: Elsevier B; 2013. [ Links ]
7. Angemi JA, Zuccotti JC. Síndrome de Joubert: a propósito de cuatro hermanos adultos afectados. Revista de Neurología. 2012; 54: 609-12. [ Links ]
8. Basson MA, Wingate R. Congenital hypoplasia of the cerebellum: developmental causes and behavioral consequences. Frontiers in Neuroanatomy. 2013; 7: 29. [ Links ]
9. Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010; 5: 20. [ Links ]
10. McGraw P. The molar tooth sign. Radiology. 2003; 229(3):671-2. [ Links ]
11. Asian H, Gongorduk K, Yildirim G, et al. Prenatal ultrasonography features of Joubert syndrome. J Clin Ultrasound. 2008; 36(9):576-580. [ Links ]
Correspondencia
Ana M. Álvarez-Sanz.
Avenida Daniel Alcides Carrión 505, Cercado, Arequipa, Perú.
Correo electrónico: amalvarezs@yahoo.es
Teléfono: 054-958910122.
Fuentes de financiamiento: Los autores declaran que la revisión fue autofinanciada.
Conflictos de interés: Los autores declaran no tener conflictos de interés relacionados con el tema de la revisión.
Recibido: 22/07/2016
Aceptado: 28/09/2016
