










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Neuro-Psiquiatría
Print version ISSN 0034-8597
Rev Neuropsiquiatr vol.79 no.3 Lima July 2016
REPORTE DE CASO
Síndrome de Kinsbourne por neuroblastoma de localización mediastinal: A propósito de un caso infantil.
Kinsbourne syndrome due to neuroblastoma of mediastinal location: A child case report.
Elizabeth R. Espíritu-Rojas1,a;2, Rosa Alvarado1,a;2, Tania Juárez1,b, Susan Samalvides1,b, Iván O. Espinoza1,b;2,d, Judith R. Vila1,b, Daniel Guillén-Pinto1,b,f;2,e
1 Hospital Nacional Cayetano Heredia. Lima, Perú.
2 Facultad de Medicina Alberto Hurtado, Universidad Peruana Cayetano Heredia. Lima, Perú.
3 Instituto Nacional de Enfermedades Neoplásicas. Lima, Perú.
a Residente de Neurología Pediátrica; b Neurólogo Pediatra; c Patólogo; d Profesor Auxiliar; e Profesor Principal; f Jefe de Servicio.
RESUMEN
Se presenta el caso de un niño de un año de edad con neuroblastoma de localización en el mediastino posterior, que debutó con manifestaciones de Síndrome de Kinsbourne (opsoclonus, mioclonus), y que mejoró ostensiblemente después de la extracción quirúrgica del tumor. Se discute la frecuencia, localización, forma de presentación y tratamiento del síndrome.
PALABRAS CLAVE: Síndrome de Kinsbourne, niño, neuroblastoma, tórax.
SUMMARY
The case of a 1-year old child with a neuroblastoma of posterior mediastinal location is presented. The initial manifestations were those of the Kinsbourne Syndrome (opsoclonus, myoclonus), which improved significantly after the surgical removal of the tumor. The frequency, location, form of presentation and treatment of the Kinsbourne Syndrome are discussed.
KEYS WORDS: Child, neuroblastoma, thorax, Kinsbourne syndrome.
INTRODUCCIÓN
El Síndrome de Kinsbourne, conocido también como Síndrome de Opsoclonus mioclonus, fue descrito por primera vez por Marcel Kinsbourne en 1962, es una rara enfermedad neurológica que afecta a niños previamente sanos, se caracteriza por opsoclonus, mioclonías y ataxia. Puede ser considerado un síndrome paraneoplásico por la asociación con neuroblastoma y otros tumores.
Se presenta el caso de un niño con una forma atípica de Síndrome de Kinsbourne por neuroblastoma de localización torácica.
Caso clínico
Paciente varón de 1 año 2 meses de edad procedente de Sayán, provincia de Huaura Lima, Perú. Acude al Hospital Nacional Cayetano Heredia (HNCH), por emergencia, con un mes de enfermedad, caracterizada por síntomas progresivos. El primer día presentó rinorrea, tos y desequilibrio, ocasionándole dos caídas, al segundo día temblor generalizado. Durante la primera semana continuó atáxico, tanto que limita la deambulación. La segunda semana no podía fijar la mirada ni realizar seguimiento visual. La tercera semana se sumó movimientos oculares involuntarios en varias direcciones muy erráticos, parpadeo constante, despertares frecuentes, así como sacudidas en las cuatro extremidades, sobresaltos, movimientos gestuales de la cara, irritabilidad constante e hiporexia. Al ingreso presentó frecuencia cardíaca de 121 por minuto, frecuencia respiratoria de 38 por minuto y presión arterial de 94/56 mmHg (Percentil 90). El paciente lucía pálido, en regular estado general, adelgazado, con dificultad respiratoria moderada. En el examen neurológico se encontraba despierto, se evidenció opsoclonus multidireccional de gran intensidad, además de flutter palpebral constante, mioclonías generalizadas a predominio de cara y extremidades que se incrementaban con los cambios de postura, llanto y movimientos, temblor generalizado simétrico de gran intensidad en reposo y acción, dismetría, ataxia de tronco, hipotonía generalizada e hiperreflexia. El resto de la evaluación dentro de la normalidad.
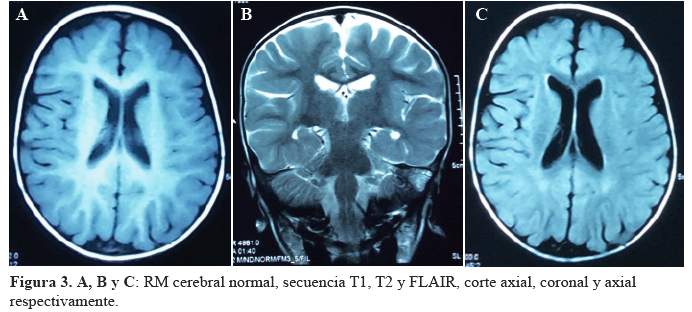
La biometría hemática mostró 10070 leucocitos por ml, hemoglobina 10,7 gr/dl y plaquetas normales. Exámenes de bioquímica (glucosa, urea, creatinina, electrolitos), perfil lipídico, y examen de orina normales, pruebas de BK negativos. En la radiografía de tórax se encontró patrón intersticial bilateral y una masa a nivel de mediastino posterior (Figura 1), y en la resonancia magnética (RM) torácica se observó unalesión delimitada, de aspecto nodular proliferativo situada en la pared postero-medial del hemitórax derecho, con un diámetro máximo de 36 mm en el eje cráneo-caudal, de señal intermedia en secuencia TI y alta en T2, con realce a la administración de contraste, sugestiva de neuroblastoma (Figura 2), resto de estructuras sin alteraciones. La RM cerebral no mostró lesión estructural (Figura 3). El electroencefalograma(EEG) fue normal. Con estos resultados se inició prednisona a 2mg/kg/día.
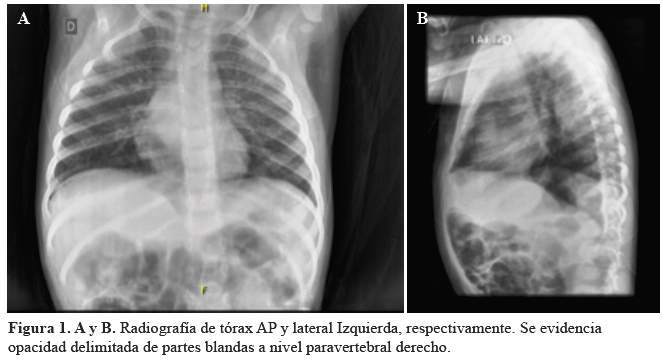
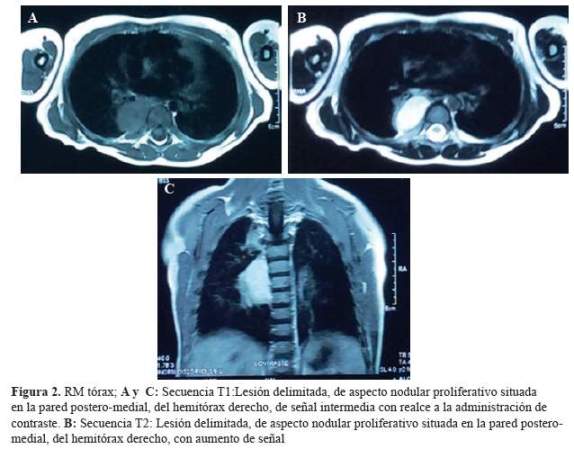
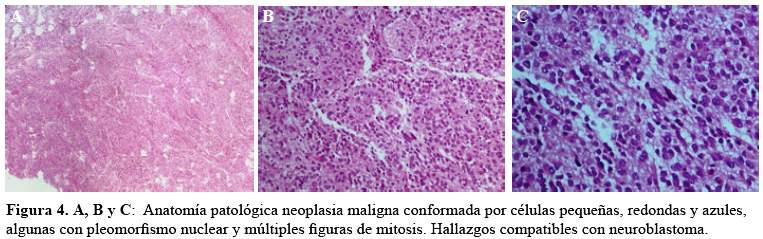
Durante dos semanas se observó una mejoría discreta con el tratamiento corticoide. A los 18 días de la hospitalización se le extirpó sin complicaciones quirúrgicas la tumoración paravertebral que se encontró adherida a la pleura parietal derecha, de bordes definidos y resecables. En los días post operatorios la mejoría fue rápidamente evidente. Paciente fue referido al Instituto Nacional de Enfermedades Neoplásicas (INEN), donde se realizó la inmunohistoquímica, describiendo neuroblastoma rico en estroma schwanniano, sinaptofisina y neurofilamento (+), KI-67 (índice proliferativo) 60%, proteina S100 (Figura 4) negativo. Se realizó aspirado de médula ósea, biopsia de hueso que fueron normales y análisis de LCR que fue también normal. Otros análisis bioquímicos y serológicos complementarios fueron normales. La nueva tomografía espiral multicorte (TEM) de cerebro y cuello sin hallazgos significativos.
En la TEM de tórax se observó engrosamiento de la pleura parietal en el tercio medio del hemitórax derecho (HTD) con extensión a partes blandas, gammagrafía ósea con captación anormal en pared de HTD, sin amplificación del gen N-MYC, siendo catalogado como Neuroblastoma Primario de mediastino posterior de bajo riesgo, EC IIA. Se le inició quimioterapia con carboplatino y etopósido. Actualmente se encuentra bajo control ambulatorio.
DISCUSIÓN
El síndrome Opsoclonus-mioclonus (SOM), fuedescrito en 1962 por M. Kinsbourne quien reportó seis niños con enfermedad neurológica denominándola encefalopatía mioclónica (1), la cual posteriormente fuedenominada “síndrome de ojos danzantes” o “Síndrome de Kinsbourne”. Es una entidad poco frecuente, que se caracteriza por tres síntomas principales, opsoclonus, mioclonus y ataxia (1-4).
El Opsoclonus es un disturbio involuntario demovimientos oculares conjugados, no fásicos, rápidos y multidireccionales, con una frecuencia de entre 6 y 15 Hz, que persiste durante el sueño. Las mioclonías son espasmos musculares irregulares que se pueden observar principalmente en los músculos del tronco, extremidades y cara. La ataxia es definida como un trastorno del equilibrio, incluye el tronco, extremidades y puede ser incapacitante en la fase aguda (1,3).
Estos síntomas a menudo se asocian con trastornos de sueño e irritabilidad, además regresión del desarrollo (1-3,5). Por lo general, se produce en los primeros 2 años de edad (5), en el caso de nuestro paciente inició al año de edad.
El SOM se asocia a infecciones y tumores derivados de la cresta neural, siendo el más frecuente el neuroblastoma (NB), referido hasta en el 50% de los casos (4,5), mucho más raro con otros tipos de cáncer (2). De otro lado, alrededor del 2 – 3 % de los pacientes con neuroblastoma (NB) presentan SOM paraneoplásico (5). Éstos pacientes suelen tener mejor pronóstico y mayor tasa de supervivencia, sin embargo, la mayoría cursa con retraso del desarrollo y disfunción neurológica a largo plazo (4, 6), en un estudio realizado por Klein et al., se encontró que también los trastornos del comportamiento y problemas cognitivos fueron los más frecuentes.
Actualmente el blanco de investigación es el desarrollo neuropsicológico de éstos pacientes ya que son muy irritables, muestran alteraciones graves de aprendizaje y pueden desarrollar retraso mental (1,5). En el caso de nuestro paciente la regresión en los hitos del desarrollo fue evidente desde su ingreso, sin embargo, los resultados a largo plazo aún son inciertos.
Según el consenso internacional llevado a cabo el 2004 en Italia la presentación aguda de SOM requiere que tres de los cuatro criterios diagnósticos siguientes estén presentes para describir el síndrome típico: 1) opsoclonus, 2) mioclonía/ataxia, 3) cambio de comportamiento y / o alteración del sueño, y 4) neuroblastoma (4,7), en nuestro reporte se cumplieron todos los criterios.
En cuanto a criterios clínicos de gravedad de los pacientes con SOM que modifica Krug et al., el año 2009(2,4) nuestro paciente presentó grado moderado a severo, lo que ameritó una conducta rápida de tratamiento.
En cuanto al mecanismo inmunológico debemos de tener en cuenta el concepto de degeneración neurológica paraneoplásica (DNP) la cual se desarrolla en asociación con el cáncer, infecciones virales o exposición a metabolitos tóxicos. Las DNPs son provocadas por una respuesta inmune anti-tumor, contra los antígenos neuronales compartidos por las células cancerosas y las neuronas normales, que conducen a la enfermedad neurodegenerativa autoinmune, siendo el resultado final producto de: (i) Apoptosis de células tumorales (ii) internalización de las células tumorales apoptóticas por células dendríticas, que migran a los ganglios linfáticos y activan las células CD4 + y T CD8 +, (iii) cruce de la BHE por las células T CD8 + activadas, dirigido contra las neuronas normales que expresan los mismos antígenos que se encuentran en las células tumorales. Estos eventos culminan con los signos clínicos de la degeneración neuronal. La etiología del SOM todavía no está clara, sin embargo, se describe autoanticuerpos en pacientes adultos y pediátricos con SOM, que tienen origen autoinmune, y en aquellos relacionados con NB, los autoanticuerpos séricos contra las neuronas y las células de Purkinje del cerebelo se han detectado consistentemente. Recientemente Panzer et al. Identificó y caracterizó anticuerpos contra el antígeno de superficie en dendritas neuronales en cerebelo y tronco cerebral en 4 de 42 sujetos con SOM (3,8).
Se han hallado autoanticuerpos IgG en suero en niños con NB y SOM, que específicamente se unen al antígeno de superficie intracelular de líneas celulares de NB, inhibiendo su proliferación, e induciendo apoptosis (9). También se ha demostrado una activación pronunciada de células B en el LCR de niños con SOM, en conjunto, estos estudios llevaron a la hipótesis de la aparición de una autoinmunidad de reacción cruzada entre NB y el sistema nervioso central (SNC) en SOM. La respuesta inmune generada por los antígenos asociados a tumores y dirigida contra las mismas moléculas expresadas por el tejido sano puede causar daño neurológico a largo plazo, pero al mismo tiempo puede tener en cuenta el pronóstico favorable del tumor (3,9).
El neuroblastoma (NB) es el tumor sólido extracraneal más frecuente en la infancia (3, 10-12), representa aproximadamente el 7-10% de los cánceres pediátricos y el 15% de todas las muertes por cáncer pediátrico en pacientes menores de 15 años de edad (13). En el 2010 la incidencia ajustada a la edad en Estados Unidos fue de 10.7 casos por 1 000 000 de personas de 0 a 14 años(12).
Es una enfermedad heterogénea, tanto biológica como clínicamente. Se postula que es un tumor embrionario que se deriva de las células precursoras del sistema nervioso (SN) periférico (simpático), el cual puede surgir en cualquier punto de la cadena simpática, (12,13), sin embargo cuando se asocia al SOM son más a menudo torácicos (mediastinal 49-61%), pero pueden originarse en el retroperitoneo abdominal (13%), adrenal (13%), sacrococcígea (1%)(14), en el caso de nuestro paciente la localización fue en mediastino posterior, si bien los tumores neurogénicos son la causa más común de masa mediastinal posterior hasta en el 90% de los casos(10), en el SOM asociado a NB la localización en el compartimento posterior del mediastino es infrecuente (15).
Según el sistema de clasificación de la International Neuroblastoma Stading System (INSS) (16) nuestro paciente fue clasificado como un tumor primario en mediastino posterior en estadío IIA, siendo de bajo riesgo con N-MYC no amplificado. De otro lado, según la última clasificación de la International Neuroblastoma Risk Group Stading System (INRGSS), se correspondería con estadío L1(17), es decir tumor localizado que no compromete estructuras vitales confinado a un compartimento del cuerpo(18).
En cuanto a su comportamiento clínico varía de regresión espontánea a progresión rápida y potencialmente mortal. Los planes de tratamiento varían e incluyen cirugía, quimioterapia y radioterapia. Para tumores en etapas I y II del INSS, la extirpación quirúrgica completa por sí sola puede proporcionar un tratamiento adecuado para una cura. Sin embargo, en pacientes con INSS estadios III o IV, la extirpación completa no siempre es factible, y en ocasiones puede causar complicaciones graves (11). En nuestro paciente se realizó extirpación quirúrgica completa, sin embargo, se encontró captación anormal en la pared del HTD mediante la gammagrafía, pautándose quimioterapia.
En cuanto a las pruebas moleculares los protooncogenes representan un grupo de genes eucariotas cuyas formas activadas están implicadas en el desarrollo de cáncer. La familia de protooncogenes MYC (C-myc, N-myc y L-myc) se relaciona con el origen de diversas neoplasias en seres humanos. El oncogen N-myc es una fosfoproteína nuclear, juega un papel importante en la tumorogénesis del neuroblastoma y define un subconjunto agresivo de los tumores. La amplificación de N-myc (definida como más de 10 copias) se encuentra en alrededor del 20% de los casos totales y confiere un pronóstico particularmente malo(12), se expresa a niveles anormalmente altos en numerosas líneas celulares de neuroblastoma (13,18), sin embargo en el caso que presentamos no hubo amplificación del gen N - myc, lo que mejora el pronóstico de nuestro paciente.
En cuanto al diagnóstico la RM es de elección para el diagnóstico de tumor primario y evaluar su extensión local, a excepción de los tumores abdominales para los que la tomografía sigue siendo la mejor opción. La extensión metastásica debe evaluarse con exploración 123 I-metaiodobenzylguanidina (MIBG), que tiene una sensibilidad entorno al 90- 95% y ecografía del hígado (2), la cual fue normal en nuestro paciente.
En cuanto al tratamiento del SOM, según la literatura revisada la primera línea de tratamiento es la corticoterapia, excepto en los que presentan remisión espontánea después de la resección quirúrgica, sin embargo existen muchas opciones terapéuticas como la inmunoterapia(2,19) , la cual será individualizada. El tiempo requerido para la reversibilidad es variable de días, semanas meses o incluso años. En nuestro caso el paciente recibió corticoterapia hasta la programación del primer ciclo de quimioterapia, se seguirá de cerca al paciente para observar curso de la enfermedad.
El receptor de quimioquinas CC tipo 7(CCR7) y sus ligandos, CCL19 y CCL21, son mediadores clave para equilibrar la tolerancia y la inmunidad en el sistema inmunológico Según un estudio realizado por Pranzatelli et al., El ligando CCL21se sobreexpresa en el suero de pacientes pediátricos con SOM y están bajo regulación de corticosteroides y ACTH, destacando la posible utilidad de CCL21 como marcador de tratamiento con esteroides (20).
En conclusión, reportamos el caso de un paciente que presentó cuadro neurológico (síndrome de Kinsbourne) como expresión de una patología tumoral primaria a nivel de mediastino posterior, de localización inusual, denominada en conjunto como un desorden neurológico paraneoplásico, el cual implica destrucción de neuronas normales que repercutirán en el neurodesarrollo a largo plazo. Recalcamos la clínica en éstos pacientes y la importancia de reconocer esta forma de presentación, siendo el tiempo un factor clave en el manejo definitivo de ésta patología y determinar así el pronóstico de éstos niños.
REFERENCIAS BIBLIOGRÁFICAS
1. Blaes F, Dharmalingam B. Childhood opsoclonusmyoclonus syndrome: Diagnosis and treatment. Expert Rev Neurother. 2016;16(6):641-8. [ Links ]
2. Krug P, Schleiermacher G, Michon J, et al. Opsoclonus-myoclonus in children associated or not with neuroblastoma. Eur J Paediatr Neurol.2010;14(5):400-9. [ Links ]
3. Raffaghello L, Conte M, De Grandis E, Pistoia V. Immunological mechanisms in opsoclonusmyoclonus associated neuroblastoma. Eur J Paediatr Neurol. 2009;13(3):219-23. [ Links ]
4. Matthay KK, Blaes F, Hero B, et al. Opsoclonus myoclonus syndrome in neuroblastoma a report from a workshop on the dancing eyes syndrome at the advances in neuroblastoma meeting in Genoa, Italy, 2004. Cancer letters.2005;228(1-2):275-82. [ Links ]
5. Klein A, Schmitt B, Boltshauser E. Long-term outcome of ten children with opsoclonus-myoclonus syndrome. Eur J Pediatr. 2007;166(4):359-63. [ Links ]
6. Mitchell WG, Brumm VL, Azen CG, Patterson KE, Aller SK, Rodriguez J. Longitudinal neurodevelopmental evaluation of children with opsoclonus-ataxia. Pediatrics. 2005;116(4):901-7. [ Links ]
7. Hero B, Schleiermacher G. Update on pediatric opsoclonus myoclonus syndrome. Neuropediatrics. 2013;44(6):324-9. [ Links ]
8. Panzer JA, Anand R, Dalmau J, Lynch DR. Antibodies to dendritic neuronal surface antigens in opsoclonus myoclonus ataxia syndrome. J Neuroimmunol. 2015; 286:86-92. [ Links ]
9. Korfei M, Fuhlhuber V, Schmidt-Woll T, Kaps M, Preissner KT, Blaes F. Functional characterisation of autoantibodies from patients with pediatric opsoclonus-myoclonus-syndrome. J Neuroimmunol. 2005; 170(1-2):150-7. [ Links ]
10. Atwal SS. Neuroblastoma of posterior mediastinum: classical imaging findings. J Clin Diagn Res. 2014; 8(10):1. [ Links ]
11. Moon SB, Park KW, Jung SE, Youn WJ. Neuroblastoma: treatment outcome after incomplete resection of primary tumors. J Pediatr Surg. 2009;25(9):789-93. [ Links ]
12. Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and therapy. Annu Rev Med. 2015; 66:49-63. [ Links ]
13. Jiang M, Stanke J, Lahti JM. The connections between neural crest development and neuroblastoma. Curr Top Dev Biol. 2011; 94:77-127. [ Links ]
14. Pranzatelli MR. The neurobiology of the opsoclonusmyoclonus syndrome. Clin Neuropharmacol. 1992; 15(3):186-228. [ Links ]
15. Solovyov AE, Morgun VV, Paholchuk AP. Neuroblastoma in pediatric patients. Klin Khir. 2015;(6):59-61. [ Links ]
16. Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106-20. [ Links ]
17. Monclair T, Brodeur GM, Ambros PF, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009;27(2):298-303. [ Links ]
18. Shohet JM. Redefining functional MYCN gene signatures in neuroblastoma. Proc Natl Acad Sci USA. 2012;109(47):19041-2. [ Links ]
19. Pranzatelli MR, Tate ED. Trends and tenets in relapsing and progressive opsoclonus-myoclonus syndrome. Brain Dev. 2016;38(5):439-48. [ Links ]
20. Pranzatelli MR, Tate ED, McGee NR, Ransohoff RM.CCR7 signaling in pediatric opsoclonus-myoclonus: upregulated serum CCL21 expression is steroidresponsive. Cytokine. 2013;64(1):331-6. [ Links ]
Declaración de autores: No tenemos conflicto de intereses.
Correspondencia
Dra. Elizabeth Ruth Espíritu Rojas
Dirección: Avenida Pacífico 180, San Miguel, Lima, Perú
Teléfono: 51943244426
Recibido: 15/08/2016
Aceptado: 19/09/2016
