










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Neuro-Psiquiatría
versión impresa ISSN 0034-8597
Rev Neuropsiquiatr vol.80 no.1 Lima ene. 2017
http://dx.doi.org/10.20453/rnp.v80i1.3061
10.20453/rnp.v80i1.3061
REPORTES DE CASO
Sindrome de Joubert asociado a malformación de Dandy-Walker en un paciente pediátrico:Reporte de un caso.
Joubert syndrome associated with Dandy-Walker malformation in a pediatric patient: a case report.
Ana M. Álvarez-Sanz 1,2,a, Lizeth Y. Cabanillas-Burgos 1,2,b, Ximena P. Huamani-Condori 3,c
1 Universidad Nacional de San Agustín. Arequipa, Perú.
2 Hospital Regional Honorio Delgado Espinoza. Arequipa, Perú.
3 Universidad Católica de Santa María. Arequipa, Perú.
a Neuróloga Pediatra; b Pediatra; c Estudiante de Medicina Humana.
RESUMEN
Se presenta el caso de una niña de 10 meses de edad que desde el nacimiento experimentó apnea e hiperpnea, hipotonía y nistagmus. Durante sus primeros meses de vida presentó además insuficiencia renal, retraso psicomotor e infecciones severas. Se diagnosticó de síndrome de Joubert asociado a malformación de Dandy-Walker y en la prueba de resonancia magnética se observó el típico signo de “Diente Molar” característicamente asociado al quiste. La asociación infrecuente de este tipo de malformaciones cerebrales agrava ciertamente el pronóstico del cuadro original.
PALABRAS CLAVE: Síndrome de Joubert, Malformación de Dandy-Walker.
SUMMARY
The case of a 10-month-old girl with apnea and hyperpnea, hypotonia and nystagmus from the time of birth, is presented. During her first months of life she presented additional manifestations of renal failure, psychomotor retardation and severe infections. The diagnosis of Joubert syndrome associated with Dandy-Walker Malformation was formulated, and the typical “Molar Tooth” sign, characteristically associated with the cyst characteristics was detected with magnetic resonance imaging tests. The uncommon association of these brain malformations that worsen the prognosis, is discussed.
KEY WORDS: Joubert syndrome, Dandy-Walker malformation.
INTRODUCCION
El síndrome de Joubert (SJ) fue descrito en 1969 por Marie Joubert, como un trastorno clínico, genético y radiológicamente heterogéneo (1,2). Este síndrome está asociado a microcefalia, facies dismórfica, distrofia retiniana, protrusión lingual, poliquistosis renal, cardiopatía congénita, lámina tectal troncoencefálica no segmentada, agenesia del cuerpo calloso y conexiones aberrantes de sustancia blanca. La resonancia es el examen de elección para identificar el típico signo del “Diente Molar” (3,4).
Tiene una prevalencia de 1/100 000 a 1/150 000 nacidos vivos y se manifiesta desde la etapa neonatal con trastornos en el ritmo respiratorio, nistagmus, hipotonía y alteraciones en la deglución (5).
El 10% de los pacientes presentan colecciones quísticas dentro del espectro de malformaciones Dandy-Walker (MDW), que requieren cumplir los siguientes criterios, dilatación quística del cuarto ventrículo, hipoplasia o agenesia del vermis cerebeloso e hidrocefalia en algún momento del curso clínico (6). Curiosamente, mientras que el 75 al 80% de los pacientes con MDW requiere shunt para hidrocefalia, ninguno de los más de 300 casos confirmados de Joubert han tenido hidrocefalia. A continuación presentamos una paciente con esta rara forma de asociación.
Caso clínico
Lactante mujer de 10 meses de edad, natural de Arequipa y procedente de Iquipí, localidad situada a 470 msnm, capital del distrito de Río Grande, provincia de Condesuyos, departamento de Arequipa. Primera hija de padres no consanguíneos y nacida a término y con peso adecuado para la edad gestacional; los primeros 15 días fue hospitalizada por presentar periodos de apnea e hiperpnea, hipotonía y nistagmus. Se le realizó ultrasonido cerebral evidenciando hidrocefalia.
A los 6 meses de edad, ingresó por sepsis de foco respiratorio con insuficiencia renal oligúrica, requirió ventilación mecánica por dos semanas y en el proceso de evaluación se detectó poliquistosis renal bilateral demostrada por tomografía. Mejoró con el tratamiento antibiótico y de soporte.
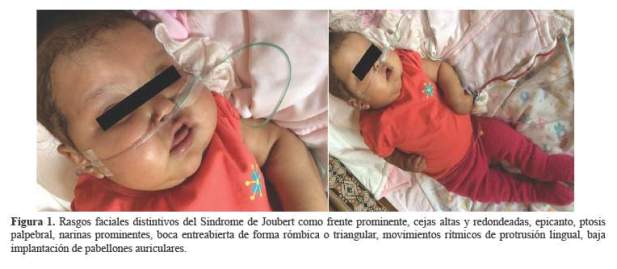
Reingresó a los 10 meses de edad, en choque séptico (foco pulmonar y ótico). Se evidencia el retraso psicomotor y las características dismórficas, con frente prominente, epicanto, ptosis palpebral, baja implantación de pabellones auriculares, boca entreabierta de forma triangular y movimientos rítmicos de protrusión lingual (figura 1). Durante esta hospitalización presentó crisis convulsivas tónicas en 3 oportunidades con duración de 10 segundos, recibiendo fenitoína por 2 semanas. Ya estable, se evidenció hipertensión arterial secundaria a la patología renal de fondo; posteriormente se complicó con neumonía intrahospitalaria, candidiasis sistémica, la anasarca y anuria requiriendo diálisis peritoneal.
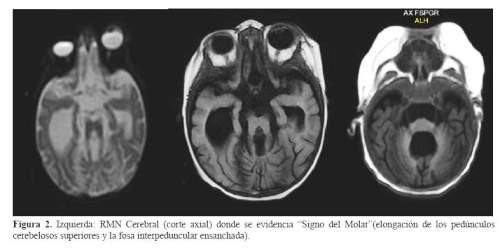
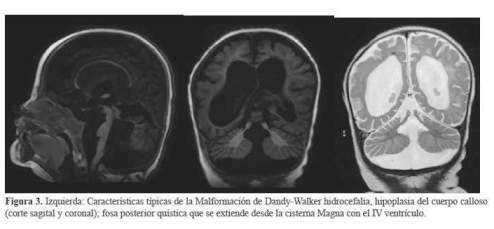
El diagnóstico de Síndrome de Joubert se confirmó con la resonancia cerebral, evidenciando hipoplasia del vermis cerebeloso, engrosamiento de los pedúnculos cerebelosos perpendiculares al tronco encefálico, fosa interpeduncular ensanchada que simulan la silueta de una muela o “Signo del diente molar”, moderada hidrocefalia, hipoplasia del cuerpo calloso, agenesia parcial del septum pellucidum, fosa posterior amplia y comunicación de cuarto ventrículo con megacisterna magna (figura 2 y figura 3).
La paciente fue transferida para continuar con diálisis peritoneal continua ambulatoria (DPCA) además del tratamiento para la hipertensión arterial,terapia física y rehabilitación.
DISCUSIÓN
El SJ es una entidad de carácter autosómica-recesiva y puede ser sospechado en lactantes con hipotonía, apnea e hiperpnea, nistagmus, apraxia oculomotora (2) y el signo del diente molar en resonancia cerebral (7).
La paciente presentó los primeros signos desde el nacimiento, meses después se evidenciaron otras características como: frente prominente, cejas altas y redondeadas, epicanto, ptosis palpebral, narinas prominentes, boca de forma triangular, movimientos rítmicos de protrusión lingual y baja implantación de pabellones auriculares (8). No presentó distrofia retiniana ni polidactilia.
Clínicamente, la niña se ubica en la variante SJ con defectos renales (no asociados a patología retiniana), variante descrita en la clasificación de SJ que incluye seis subgrupos fenotípicos (9), conocidos como SJ puro, SJ con distrofia retiniana, SJ con defectos oculorrenales, SJ con defecto hepático y SJ con defectos orofacio digitales (por ejemplo, lengua bífida, hamartomas múltiples, múltiples frenillos orales y polidactilia) (6,10,11). En todos, el hallazgo fundamental son los diferentes grados de displasia cerebelosa (12). En la niña la poliquistosis renal bilateral fue diagnosticada a los 6 meses de edad y evolucionó rápidamente a insuficiencia renal crónica tributaria de diálisis.
El diagnóstico durante la etapa fetal es posible de realizar mediante análisis moleculares y estudios de imagen como ecografía tridimensional de alta resolución y resonancia magnética fetal. El “signo del molar” está presente desde la semana 24 ± 4 de la gestación y puede ser observado por resonancia, siendo de utilidad cuando no es posible realizar el estudio molecular. En manos expertas y con equipos de última generación, son posibles de identificar algunas malformaciones de la fosa posterior como las que se presentan en la MDW desde la semana 11-13 de la gestación (11). Para el caso de la niña el ultrasonido convencional realizado durante su primera hospitalización, sólo permitió informar sobre la hidrocefalia.
Durante el desarrollo del cerebelo, específicamente del vermis y tallo encefálico, los cilios primarios regulan algunas vías embrionarias implicadas en los principales procesos del desarrollo de proliferación neuroblástica y migración axonal, incluyendo la diferenciación de las células de Purkinje y neuronas granulares (13), lo cual puede explicar la presencia de hipoplasia del vermis cerebeloso como componente de la MDW como parte de la heterogeneidad genética que comparte con el SJ.
Se han detectado hasta hoy 10 genes asociados (7), las mutaciones en estos genes están asociadas con el funcionamiento de los cilios primarios, diferentes proteínas interaccionan para formar complejos necesarios para la formación de los cilios; las múltiples funciones de estos cilios en diferentes órganos y tejidos explican por qué diversas mutaciones en estos genes pueden estar asociadas con diversas variantes clínicas.
Es importante hacer una clara delimitación radiológica entre SJ y MDW debido a las importantes diferencias que hay tanto para el manejo de pacientes, asesoramiento genético y pronóstico (6). Se ha reportado que el tamaño del istmo del tronco encefálico fue normal en sujetos con MDW, en contraste con los portadores de SJ. Además, ningún caso de MDW tenía un signo de diente molar. Estos datos sugieren dimensiones específicas en la anatomía del tronco encefálico, las cuales fueron utilizadas para distinguir radiológicamente MDW y SJ en niños con hipotonía, ataxia y retraso en el desarrollo (6). Entre los niños con SJ que tienen un quiste de fosa posterior, ninguno desarrolló hidrocefalia, si tenían elsigno del molar. Estos datos refuerzan la observación que el istmo es normal en pacientes con MDW y que el signo del molar es un marcador válido de mal funcionamiento del istmo (6). En cualquier caso, la presencia de un quiste en la fosa posterior en un niño con retraso e hipotonía deben inducir a una revisión cuidadosa del istmo para distinguir entre SJ y MDW (14).
Para el caso de la niña, las crisis epilépticas se interpretaron como secundarias al estado agudo de uremia. Aunque la epilepsia puede ser frecuente en SJ y se describen casos asociados a heterotopías cerebrales, ésta no fue considerada en la paciente ya que las crisis no se repitieron tras el retiro de la medicación antiepiléptica (15).
El neurodesarrollo y la cognición se pueden afectar en grado variable en el SJ. Los pacientes con MDW aislado sin malformaciones del sistema nervioso fuera de la fosa posterior (disgenesia del cuerpo calloso, quistes interhemisféricos, heterotopias de materia gris, meningocele occipital) u otras malformaciones sistémicas (cardiopatía defectos septal) tienen un resultado favorable. En contraste con la mayoría de los niños con SJ quienes presentan autismo y pobre desarrollo intelectual explicándose este último por la disfunción hemisférica cerebral (6,16). En otros casos, el pronóstico del SJ queda condicionado a la gravedad del cuadro respiratorio, en algunos casos las crisis de apnea remiten espontáneamente durante el primer año de vida y otras requieren asistencia mecánica. También las disfunciones hepáticas y/ o renales suelen condicionar el pronóstico vital (17).
En conclusión, la asociación entre el SJ y la MDW, es infrecuente, poco reconocida y de mal pronóstico. Se ha informado por lo menos en 10% de los casos de SJ y puede ser explicada como disgenesias que son el resultado de alteraciones de la neurogénesis temprana por mutaciones de los genes relacionados con la expresión del gradiente dorsoventral (11).
REFERENCIAS BIBLIOGRÁFICAS
1. Joubert M, Eisenring JJ, Robb JP, et al. Familial agenesis of the cerebellar vermis: A syndrome of episodic hyperpnea, abnormal eye movements,ataxia, and retardation. Neurology. 1969; 19: 813-25. [ Links ]
2. Parisi M, Glass I. Joubert syndrome and related disorders. In: Pagon R, Adam M, Bird T, Dolan C, Fong C, Sthephens K (eds.). Gene Reviews. Seattle: University of Washington; 2003. p. 1993-2013. [ Links ]
3. Calleja-Perez B, Fernández-Jaén A, Martínez-Bermejo A, Pascual-Castroviejo I. Sindrome de Joubert: presentación de cinco casos. Rev Neurol. 1998; 26 (152): 548-550. [ Links ]
4. Burguete A, Cabada T, Bacaicoa M, et al. Síndrome de Joubert: hallazgos en resonancia magnética convencional y tensor de difusión. Radiología. 2012;54(3): 279-282. [ Links ]
5. Satran D, Pierpont MEM, Dobyns WB. Cerebellooculo-renal syndromes including Arima, Senior-Loken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet. 1999; 86: 459-69. [ Links ]
6. Maria BL, Bozorgmanesh A, Kimmel KN, Theriaque D, Quisling RG. Quantitative assessment of brainstem development in Joubert Syndrome and Dandy-Walker syndrome. J Child Neurol. 2001;16(10):751-758. [ Links ]
7. Valente E, Dallapiccola B, Bertini E. Joubert syndrome and related disorders: 1879-1888. In: Dulac O, Lassonde M, Sarnat H. Handbook of clinical neurology: Pediatric Neurology Part III. Paris:Elsevier B; 2013. [ Links ]
8. Alvarez AM, Cabanillas L, Huamani X. Síndrome de Joubert. Rev Neuropsiquiatr. 2016; 79(3): 169-171. [ Links ]
9. Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010; 5: 20. [ Links ]
10. Angemi JA, Zuccotti JC. Síndrome de Joubert: a propósito de cuatro hermanos adultos afectados. Revista de Neurología. 2012; 54: 609-12. [ Links ]
11. Peña-Landín D, Carmona-Vázquez C, Medina-Crespo V, et al. Joubert-Boltshauser Syndrome associated to Dandy-Walker malformation: Report of two cases. Rev Mex Neuroci. 2014; 15(2): 112-118. [ Links ]
12. McGraw P. The molar tooth sign. Radiology. 2003;229(3):671-2. [ Links ]
13. Basson MA, Wingate R. Congenital hypoplasia of the cerebellum: developmental causes and behavioral consequences. Frontiers in Neuroanatomy. 2013; 7: 29. [ Links ]
14. Spinella GM. Research directions: Follow-up of the Joubert syndrome workshop, October 21, 1998. J Child Neurol. 1999; 14(10):667-8. [ Links ]
15. Barreirinho S, Teixeira J, Moreira N, Bastos C, Gonçalves S, Barbot C. Síndrome de Joubert: revisión de 12 casos. Rev Neurol. 2001; 32 (9): 812-817. [ Links ]
16. Calabro F, Arcur T, Jinkins JR. Blake’s pouch cyst: An entity within the Dandy-Walker continuum. Neuroradiology. 2000; 42: 290-295. [ Links ]
17. Asian H, Gongorduk K, Yildirim G, et al. Prenatal ultrasonography features of Joubert syndrome. J Clin Ultrasound. 2008; 36(9): 576-580. [ Links ]
Fuentes de financiamiento: Los autores declaran que el estudio fue autofinanciado.
Conflictos de interés: Los autores declaran no tenerconflictos de interés con respecto al caso clínico presentado.
Correspondencia
Ana M. Álvarez-Sanz.
Avenida Daniel Alcides Carrión 505, Cercado, Arequipa, Perú.
Correo electrónico: amalvarezs@yahoo.es
Teléfono: 51958910122
Recibido: 11/12/2016
Aceptado: 06/02/2017
