










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Neuro-Psiquiatría
Print version ISSN 0034-8597
Rev Neuropsiquiatr vol.80 no.4 Lima Oct./Dec. 2017
http://dx.doi.org/https://doi.org/10.20453/rnp.v80i4.3242
REPORTE DE CASO
Síndrome de Miller Fisher en un niño de 12 años
Miller Fisher Syndrome in a 12 year old boy
Cynthia L. Samaniego – Lozano 1,a;2 , Tania Juárez 1,b, Iván O. Espinoza 1,b;2, Judith Vila 1,b, Daniel Guillén- Pinto 1,b,c;2.
1 Hospital Cayetano Heredia. Lima, Perú.
2 Facultad de Medicina Alberto Hurtado, Universidad Peruana Cayetano Heredia. Lima, Perú.
a Residente de Neurología Pediátrica; b Neurólogo Pediatra; c Jefe del Servicio de Pediatría.
RESUMEN
El Síndrome de Miller Fisher (SMF) es una variante del Síndrome de Guillain Barré (SGB), caracterizado por la tríada clínica de oftalmoplejía, ataxia y areflexia. Se presenta el caso de un niño de 12 años de edad, examinado con un tiempo de enfermedad de 4 días y con una variedad de síntomas que incluían ptosis palpebral, somnolencia, marcha tambaleante y debilidad muscular, asociados a antecedente de infección respiratoria de vías altas. El examen clínico demostró paresia del III, IV, y VI nervios craneales de ambos ojos, arreflexia y debilidad distal en extremidades. Se instaló tratamiento con Inmunoglobulina intravenosa que condujo a una evolución clínica satisfactoria.
PALABRAS CLAVE: Anticuerpos anti-GQ1b, ataxia, arreflexia, síndrome de Miller-Fisher.
SUMMARY
The Miller Fisher Syndrome (MFS) is a variant of the Guillain Barre Syndrome (GBS), characterized by the clinical trial of ophthalmoplegia, ataxia and areflexia. The case of a 12 year old boy is examined with a 4-day long history characterized by symptoms such as palpebral ptosis, somnolence, ataxia and muscle weakness, associated with a history of upper respiratory infection. Clinical examination showed paresis of III, IV, and VI cranial nerves of both eyes, areflexia, and distal weakness in the extremities. Treatment with intravenous immunoglobulin was established, leading to a satisfactory clinical evolution.
KEYWORDS: Anti-GQ1b antibody ataxia, areflexia, Miller-Fisher syndrome.
INTRODUCCIÓN
El Síndrome de Miller Fisher (SMF) es una variante poco frecuente del Síndrome de Guillain- Barré (SGB), caracterizada por la tríada clínica de oftalmoplejía, ataxia y arreflexia (1). Esta descripción fue descrita por primera vez en 1932 y desde entonces se ha reportado en todas las latitudes.
Presentamos un caso de SMF en un niño, con la finalidad de difundir las características clínicas particulares en esta edad, de manera que permita reconocer y tratar esta enfermedad en forma adecuada y oportuna, para evitar complicaciones mayores, como la insuficiencia respiratoria.
Caso clínico
Paciente varón de 12 años, que acude a emergencia por enfermedad de 4 días que inició con somnolencia y caída palpebral derecha. En los dos días siguientes continuó somnoliento, manteniendo la caída palpebral y agregándose marcha en zigzag y debilidad en piernas, que impedía caminar sin apoyo. Al cuarto día, la debilidad progresa, comprometiendo los brazos, y la inestabilidad se acentúa, impidiendo la bipedestación, la caída palpebral afectó ambos ojos y el niño comenzó a tener visión doble horizontal. Tres días antes, el niño había tenido una infección respiratoria de vías altas.
Al examen, presentaba funciones vitales estables: frecuencia cardíaca 90/minuto, frecuencia respiratoria 13/minuto, presión arterial de 100/56 mmHg (percentil 50), y saturación de oxígeno en 93%. Tenía patrón respiratorio regular, auscultación cardiopulmonar normal, abdomen depresible y sin organomegalias. Al examen neurológico estaba lúcido y orientado, con debilidad simétrica y distal de grado 4/5 en las cuatro extremidades, sensibilidad normal, arreflexia osteotendinosa en las extremidades, ptosis palpebral parcial bilateral, debilidad de los músculos extraoculares rectos medial, superior, inferior, lateral y oblicuos de ambos ojos, pupilas redondas, iguales que reaccionaban lentamente a la luz, fondo de ojo normal bilateral. Con funciones superiores conservadas. Marcha y signo de Romberg no valorable porque la inestabilidad postural del paciente no permitía la bipedestación (figura 1).
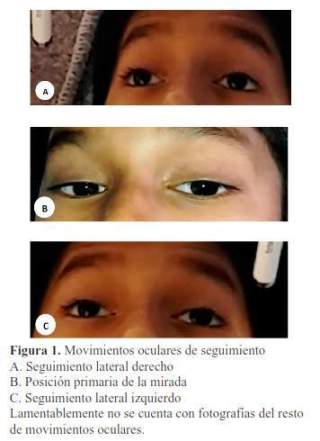
El primer día de la hospitalización, se le realizó una primera electromiografía (EMG) y velocidad de conducción nerviosa (VCN), demostrándose ausencia de reflejo H y una segunda electromiografía realizada al inicio de la tercera semana de enfermedad, que demostró actividad de inserción incrementada en parte distal de miembros inferiores, escasas unidades motoras polifásicas en ambos tibiales anteriores con reclutamiento disminuido, compatible con polineuropatía axonal motora.
Entre el primer y segundo día de hospitalización aumenta la dificultad respiratoria y se agrega disfagia y disartria, pasando a cuidados intensivos para monitorización. Al tercer día de hospitalización se inició el tratamiento con inmunoglobulina humana (IVIG) a una dosis de 400 mg/kg/día administrada durante 5 días. A las 24 horas de tratamiento se observó mejoría en el nivel de conciencia y una semana después mejoró la debilidad de los nervios oculomotores, logró sentarse con apoyo y reaparecieron los reflejos osteotendinosos en los brazos. Posteriormente, a la segunda semana logró caminar con asistencia de otra persona. Hasta que a los 28 días salió de alta, valiéndose de apoyo mínimo. Un mes después era capaz de caminar por lo menos tres metros sin ayuda, aun con ptosis palpebral bilateral y oftalmoparesia.
Los análisis iniciales revelaron hemoglobina 13,7 g/dL, leucocitos 7980/mm3, bastones 0%, segmentados 67%, eosinofilos 4,6%, linfocitos 21%, plaquetas 410000/mm3, urea 20,1, creatinina 0,4, sodio 142, potasio 4,35, cloro 102, la prueba de antígeno fecal para Campylobacter (ELISA) fue negativa. El análisis del líquido cefalorraquídeo (LCR), mostró proteínas 256 mg / dL, glucosa 55 mg/dL, glóbulos blancos 4 células/ml, glóbulos rojos 6 /ml, coloración Gram negativo y cultivo negativo. La prueba para los anticuerpos anti-GQ1b fue negativa y la resonancia magnética de encéfalo no presentó alteraciones.
DISCUSIÓN
El Síndrome de Miller Fisher (SMF) se caracteriza por la tríada clínica de oftalmoplejía, ataxia y arreflexia. Esta triada fue descrita por primera vez por James Collier en 1932 y posteriormente en 1956 Charles Miller Fisher, la divulgó como una variante limitada del Síndrome de Guillain-Barré (SGB)(1). Representa aproximadamente 1% al 5% de todos los casos de SGB en los países occidentales y hasta 19% y 25% en Taiwán y Japón respectivamente (2). Existe un predominio masculino establecido en una proporción de 2:1 y una edad media de inicio de 43,6 años, aunque se han notificado casos de MFS en todos los rangos de edad (3), los reportes en niños menores de 15 años son escasos, no habiéndose notificado casos en Perú.
La patogénesis de la enfermedad se inicia con una respuesta inmune aberrante dirigida contra el tejido nervioso, por un mecanismo de autoinmunidad llamado mimetismo molecular, es decir, anticuerpos o células T activadas en respuesta a un agente infeccioso podrían reaccionar de forma cruzada contra antígenos propios, en concreto un glucoesfingolípido, el gangliósido GQ1b(4). Los agentes infecciosos desencadenantes, más frecuentes son Campylobacter jejuni (C. jejuni) y citomegalovirus (CMV), aunque también se detectan infecciones por el virus de Epstein Barr, VIH, Mycoplasma pneumoniae, Haemophilus influenzae, virus de la hepatitis, entre otros (5).
El tiempo medio de aparición de los síntomas neurológicos posterior a la infección se refiere entre 8-10 días (rango de 1-30), rara vez se asocia al proceso en su fase aguda. En el caso presentado se observa que los síntomas neurológicos aparecieron casi terminando la infección respiratoria aguda hasta alcanzar un nadir clínico a la semana de evolución (rango de 2-21) después de los síntomas neurológicos iniciales (2). Esta evolución rápida se reporta en la mayoría de los casos reportados en niños (6).
Se ha propuesto que los síntomas neurológicos dependen de la localización específica del gangliósido y su interacción con el anticuerpo (7). Así por ejemplo, la oftalmoplejía es el resultado, de la unión de los anticuerpos antigangliósidos tipo IgG con los gangliósidos GQ1b, que se encuentran en gran proporción en las regiones paranodales de la porción infranuclear de los nervios oculomotores, troclear y abducens (8),resultando en una alteración de la motilidad extraocular (9).Además se describe que la oftalmoplejía interna, podría ser secundaria a la unión de los anticuerpos anti GQ-1b al ganglio ciliar, produciendo la denervación del esfínter pupilar (10). Nuestro paciente presentó inicialmente compromiso del III nervio craneal, con ptosis palpebral unilateral que posteriormente fue bilateral y cuando ingresa a emergencia ya presentaba limitación de los movimientos oculares.
Las pupilas lentamente reactivas a la luz, indican que hubo afección de las fibras pupilomotoras. Estos trastornos pupilares en el SMF pueden observarse hasta en el 50% de los casos, tienden a progresar independientemente de la oftalmoplejía externa, y habitualmente suelen resolverse en forma precoz (2). Del mismo modo, la ataxia, se relaciona con la unión del anticuerpo IgGantiGQ1b a las neuronas del ganglio de la raíz dorsal y al tracto espinocerebeloso (11), también se postula una disfunción del sistema aferente propioceptivo. En el caso de nuestro paciente presentó una ataxia de grado incapacitante, que lo confinó a la cama, a pesar de no tener una debilidad muscular importante al inicio, este tipo de ataxia, típicamente se describe en el SMF (6). Respecto a la arreflexia, se propone que el sitio de lesión sería a nivel de los husos musculares (12) y también por afección del sistema aferente propioceptivo (13).
El diagnóstico de SMF se realiza por las características clínicas y el curso de la enfermedad. Sin embargo, el anticuerpo anti-GQ1b, es utilizado como un marcador diagnóstico, ya que se relaciona con la actividad de la enfermedad, pero no es exclusivo del SMF, se ha identificado en otras condiciones resultando en lo que algunos expertos han designado como un “síndrome de anticuerpo anti-GQ1b”. El SMF, la oftalmoparesia aguda sin ataxia (OA), la encefalitis de Bickerstaff (EB) y el SGB con oftalmoplejía, serían parte del espectro continuo de este síndrome (14). Nuestro paciente tuvo un dosaje negativo de anticuerpo anti-GQ1b, este resultado no descarta la enfermedad, ya que según los datos reportados, el anticuerpo resulta positivo solo en alrededor del 90% de los casos (15,16), además una posible explicación a este resultado negativo, sería una baja concentración de anticuerpos anti-GQ1b al momento de la toma de muestra (tercera semana de enfermedad), los cuales tiene tienen un pico al inicio de la clínica y disminuyen hasta desaparecer en unas 4-5 semanas (17).
Los hallazgos de electromiografía y velocidad de conducción nerviosa del paciente, coinciden con lo que se describe en la literatura, típicamente, el reflejo H del sóleo está ausente, producto de la afectación de los aferentes IA de los husos musculares y durante el curso de la enfermedad, aproximadamente el 15% de los pacientes con SMF sufre una superposición de SGB axonal con anomalías en la conducción nerviosa que reflejan la disfunción axonal (13).
Respecto al tratamiento, existen pocos estudios en niños, nosotros decidimos tratar al paciente en vista de la afección clínica grave, particularmente la insuficiencia respiratoria. Seguimos las directrices de la Academia Americana de Neurología (AAN), que recomiendan terapia inmunomoduladora, con inmunoglobulina intravenosa (IVIG) y plasmaféresis (18). En el caso del paciente se prefirió la terapia con inmunoglobulina intravenosa (IVIG),debido a la relativa seguridad y facilidad de administración, aunque no hay datos confiables que sugieran sea superior a la plasmaféresis, pequeños ensayos aleatorios disponibles en los niños sugieren que IVIG acorta el tiempo de recuperación en comparación con sólo el tratamiento de sostén (19,20). Llama la atención la rápida mejoría de la motilidad ocular luego del inicio del tratamiento con IVIG con un tiempo de respuesta mucho más corto a lo descrito en la literatura. No obstante, la recuperación de la fuerza y la marcha, se observaron dentro de lo habitual.
Si bien nuestro paciente inició con un SMF típico la evolución posterior con compromiso respiratorio y debilidad de extremidades que impiden la deambulación a pesar de la desaparición de la ataxia, así como los hallazgos de la segunda EMG indican la superposición del SMF con la presentación sistémica axonal del SGB hecho que también está descrito en la literatura (21).
Con este reporte de caso estamos contribuyendo con el conocimiento de esta enfermedad en niños de nuestro medio, condición de presentación infrecuente, pero típica, la que requiere un rápido reconocimiento y manejo oportuno.
REFERENCIAS BIBLIOGRAFICAS
1. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and arreflexia). N Engl J Med. 1956; 255: 57-65. [ Links ]
2. Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher syndrome. Neurology. 2001, 56(8):1104-1106. [ Links ]
3. Snyder LA, Rismondo V, Miller NR. The Fisher Variant of Guillain-Barre Syndrome (Fisher Syndrome). J Neuro-Opthalmol. 2009; 29(4): 312–324. [ Links ]
4. Chiba A, Kusunoki S, Shimizu T, Kanazawa I. Serum IgG antibody to ganglioside GQ1b is a possible marker of Miller Fisher syndrome. Ann Neurol. 1992; 31(6): 677-679. [ Links ]
5. Ortega MG, Jaramillo G, Ancer J, Trujillo JR. Mimetismo molecular en la neuropatogénesis del síndrome de Guillain-Barré. RevMexNeuroci. 2005;6(5): 440-447. [ Links ]
6. Becker W, Watters G, Humphreys P. Fisher syndrome in childhood. Neurology. 1981;31(5): 555-558. [ Links ]
7. Kusunoki S. Diagnosis, pathogenesis and treatment of Miller Fisher syndrome and related disorders: clinical significance of antiGQ1b IgG antibody. Expert Rev Neurother.2003; 3(1): 133–140. [ Links ]
8. Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: Clinical and immunohistochemical studies. Neurology. 1993;43(10): 1911-1917. [ Links ]
9. Blanco-Marchite C, Buznego-Suárez L, Fagúndez-Vargas M, Méndez-Llatas M, Pozo-Martos P. Síndrome de Miller Fisher, oftalmoplejía interna y externa tras vacunación antigripal. Arch Soc Esp Oftalmol. 2008; 83(7): 433-436. [ Links ]
10. Radziwill A, Steck A, Borruat F, Bogousslavsky J. Isolated internal ophthalmoplegia associated with IgG anti-GQ1b antibody. Neurology. 1998; 50(1): 307. [ Links ]
11.Kusunoki S, Chiba A, Kanazawa I. Anti-GQ1b antibody is associated with ataxia as well as ophthalmoplegia. Muscle Nerve. 1999; 22(8): 1071-1074. [ Links ]
12. Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry. 2013;84(5): 576-583. [ Links ]
13. Kuwabara S, Sekiguchi Y, Misawa S. Electrophysiology in Fisher syndrome. Clinical Neurophysiology. 2017; 128(1): 215-219. [ Links ]
14. Odaka M, Yuki N, Hirata K. Anti-GQ1b IgG antibody syndrome: clinical and immunological range. Neurol Neurosurg Psychiatry. 2001; 70(1):50–55. [ Links ]
15. Willison H, Veitch J, Paterson G, Kennedy P. Miller Fisher syndrome is associated with serum antibodies to GQ1b ganglioside. J Neurol Neurosurg Psychiatry. 1993; 56(2): 204–206. [ Links ]
16. Koga M, Yuki N, Tai T, Hirata K. Miller Fisher syndrome and Haemophilus influenzae infection. Neurology. 2001; 57(4):686-691. [ Links ]
17. Rojas R, Gallardo E, Serrano C, et al. Anticuerpos anti- GQ1b: utilidad de su determinación en el diagnóstico del síndrome de Miller-Fisher. Med Clin. 2001; 116(20):761-764. [ Links ]
18. Hughes RA, Wijdicks EF, Barohn R, et al. Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003; 61(6):736-740. [ Links ]
19. Gurses N, Uysal S, Cetinkaya F, Islek I, Kalayci A. Intravenous immunoglobulin treatment in children with Guillain-Barré syndrome. Scand J Infect Dis. 1995; 27(3):241-243. [ Links ]
20. Korinthenberg R, Schessl J, Kirschner J, Mönting J. Intravenously administered immunoglobulin in the treatment of childhood Guillain-Barré syndrome: a randomized trial. Pediatrics. 2005;116(1):8-14. [ Links ]
21. Dimachkie MM, Barohn RJ. Guillain-Barré syndrome and variants. Neurol Clin. 2013; 31(2): 491-510. [ Links ]
Correspondencia:
Cynthia Luz Samaniego Lozano.
Departamento de Pediatría. Hospital Cayetano Heredia.
Avenida Honorio Delgado 262. San Martín de Porres, Lima 31.
Correo electrónico: cynthia.samaniego.l@upch.pe
Recibido: 28/11/2017
Aceptado: 11/12/2017

