










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Neuro-Psiquiatría
Print version ISSN 0034-8597
Rev Neuropsiquiatr vol.80 no.4 Lima Oct./Dec. 2017
http://dx.doi.org/https://doi.org/10.20453/rnp.v80i4.3243
REPORTE DE CASO
Epilepsia por porencefalia familiar: hallazgos clínicos, electroencefalográficos y de resonancia magnética.
Familial porencephaly epilepsy: clinical, EEG and MRI findings.
Walter F. De la Cruz Ramírez 1,2,a;3,b
1 Unidad de Epilepsia, Instituto Neurológico Eskenazi, Clínica Ricardo Palma. Lima, Perú.
2 Departamento de Epilepsia, Instituto Nacional de Ciencias Neurológicas. Lima, Perú.
3 Universidad Peruana Cayetano Heredia. Lima, Perú.
a Médico-Neurólogo ; b Profesor de Farmacología.
RESUMEN
La porencefalia familiar es un trastorno genético raro que produce quistes porencefálico, los cuales son secundarios a un infarto vascular prenatal o perinatal. La hemiparesia congénita, el retardo mental y la epilepsia de grado variable son las manifestaciones más frecuentes. Se describe el caso de dos hermanos, uno varón y la otra mujer, quienes presentan hallazgos en imágenes de resonancia magnética cerebral de quistes porencefálicos extensos que comprometen las regiones fronto-parieto-temporales de un hemisferio distinto en cada uno. Ambos presentaban hemiparesia congénita, retardo mental y epilepsia medicamente tratable con hallazgos electro-clínicos, de imágenes y neuropsicológicos que permitieron localizar la zona epileptogénica sobre la región dorsolateral del lóbulo frontal yacente al quiste porencefálico. En pacientes con hemiparesia congénita, retardo mental y epilepsia con quiste porencefálico se debe considerar la posibilidad de porencefalia familiar e indagar antecedentes familiares de este trastorno.
PALABRAS CLAVE: Quiste porencefálico, porencefalia familiar, epilepsia.
SUMMARY
Familial porencephaly is a rare genetic disorder resulting in porencephalic cysts, which are secondary to prenatal or perinatal vascular infarction. Congenital hemiparesis, mental retardation, and epilepsy in variable degrees are the most frequent manifestations. We describe the case of two siblings, one male and the other female, who present findings in brain magnetic resonance imaging of extensive porencephalic cysts that compromise the fronto-parietal- temporal regions of a different hemisphere in each. Both presented congenital hemiparesis, mental retardation and medically treatable epilepsy with electro-clinical, imaging and neuropsychological findings allowed to locate the epileptogenic zone on the dorsolateral region of the frontal lobe lying to the porencephalic cyst. In patients with congenital hemiparesis, mental retardation and epilepsy with porencephalic cyst, the possibility of familial porencephaly should be considered and a family history of this disorder should be investigated.
KEY WORDS:Porencephalic cyst, familial porencephaly, epilepsy.
INTRODUCCIÓN
La porencefalia es un trastorno extremadamente raro que produce encefalomalacia y la formación de una cavidad quística (quiste porencefálico) revestida por sustancia blanca (1). El quiste se localiza en el parénquima de un hemisferio cerebral, está lleno de líquido cefalorraquídeo y puede comunicarse con los ventrículos laterales y/o el espacio subaracnoideo (1,2).
La porencefalia es causada por una injuria encefaloclástica del parénquima cerebral secundario a un infarto vascular durante el periodo prenatal o perinatal (mayor a 24 semanas de gestación) cuya etiología en la mayoría de los casos es desconocido y en algunos puede ser genética (2). Al momento se han descrito casos de porencefalia familiar hereditaria con hemiplejía congénita atribuidos a mutaciones autosómica dominantes en el gen alfa-1 y 2 del colágeno tipo IV (COL4A1 y COL4A2) (3-5).
Las manifestaciones clínicas de la porencefalia van desde una leve hemiparesia a severa tetraplejia, epilepsia, retardo mental y cuando se trata de estructuras profundas de la línea media, defectos ópticos, hipotalámicos y pituitarios (6,7). A pesar de la conocida asociación de la porencefalia con epilepsia, la literatura sobre la semiología de las crisis epilépticas y las localización de la zona epileptogénica han sido aún poco estudias.
El objetivo de este artículo fue describir los hallazgos clínicos, electroencefalográficos y de resonancia magnética de encéfalo de dos hermanos con epilepsia por porencefalia familiar probable.
Reporte de casos
Familia
El primer miembro afectado de esta familia es la madre de siete hermanos, dos de los cuales son los casos índices que describiremos más adelante. Según relatan los hijos, la madre tenía hemiparesia desde el nacimiento y presentaba marcha hemipléjica y pulgar cautivo. Esta señora murió a los 47 años de edad por causas desconocidas por los hijos que solo refirieron que antes del fallecimiento estaba postrada y con desnutrición crónica (figura 1).
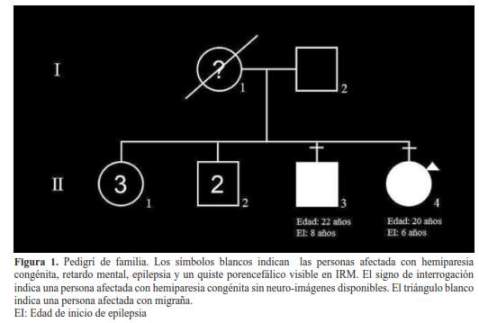
Caso 1
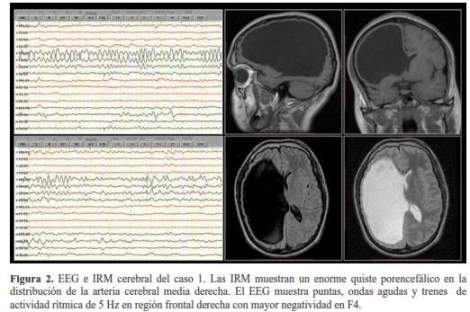
Varón de 22 años de edad, nacido a término por parto eutócico, diestro, con hemiparesia izquierda desde el nacimiento, retardo mental y epilepsia desde los 8 años de edad. Presenta crisis epilépticas de inicio focal con alteración de la conciencia caracterizadas por versión oculo-cefálica forzada hacia la izquierda seguido por postura tónica asimétrica de las extremidades por un periodo de 1 a 2 minutos que esporádicamente progresan a una crisis tónico-clónico bilateral. El paciente actualmente está libre de crisis epilépticas por más de un año tomando 600 mg/día de carbamazepina, anteriormente había tomado fenitoína y acido valproico en forma irregular y presentaba uno o dos crisis epilépticas por mes. El examen físico general y neurológico mostró desorientación parcial en espacio y tiempo, paresia e hiperreflexia osteotendinosa en hemicuerpo izquierdo. La evaluación neuropsicológica mostró compromiso neuropsicológico moderado a nivel fronto-parieto- temporal derecho con nivel intelectual extremadamente bajo para la edad, deficiencias en la asimilación y consolidación de la información predominantemente de tipo visual y alteraciones visuo-constructivas. La dominancia hemisférica cerebral para el lenguaje era izquierda. En las imágenes de resonancia magnética (IRM) de encéfalo se halló una cavidad en el hemisferio cerebral derecho, a nivel fronto-parieto-temporal, delimitado por sustancia blanca el cual se comunica con el ventrículo lateral y adelgaza el parénquima cerebral. En el electroencefalograma (EEG) estándar: se observó enlentecimiento focal continuo de la actividad cerebral sobre la región fronto-central derecha con presencia de puntas, ondas agudas y ráfagas de actividad rítmica de 5 Hz en región frontal derecha con mayor negatividad en F4 (figura 2).
Caso 2
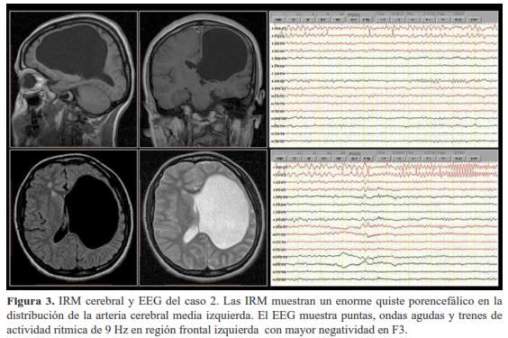
Mujer de 20 años de edad, nacida a término por parto eutócico, zurda para la escritura y cualquier praxia manual, con hemiparesia derecha desde el nacimiento, retardo mental, epilepsia desde los 6 años de edad y cefaleas episódicas con características de migraña desde la adolescencia. Presenta crisis epilépticas de inicio focal con alteración de conciencia que inicia con gemido y mirada fija por unos breves segundos y que rápidamente evoluciona a una crisis tónico-clónica bilateral que duran 1 a 2 minutos y son seguidas por somnolencia, parestesias en hemicuerpo derecho y destellos móviles de colores en ambos campos visuales. La paciente actualmente está libre de crisis epilépticas por más de un año tomando 600 mg/día de carbamazepina, anteriormente había tomado fenitoína y acido valproico en forma irregular y presentaba uno a tres crisis epilépticas por mes. El examen físico general y neurológico mostró desorientación parcial en espacio y tiempo, paresia e hiperreflexia osteotendinosa en hemicuerpo derecho. La evaluación neuropsicológica mostró compromiso neuropsicológico moderado a nivel fronto-temporal izquierdo con nivel intelectual extremadamente bajo para la edad con deficiencias en la asimilación y consolidación de la información predominantemente de tipo verbal. La dominancia hemisférica cerebral para el lenguaje era derecha. En las IRM de encéfalo se halló una cavidad en el hemisferio cerebral izquierdo, a nivel fronto-parieto-temporal, delimitado por sustancia blanca el cual se comunica con el ventrículo lateral y adelgaza el parénquima cerebral. En el EEG estándarse observó: enlentecimiento focal continúo de la actividad de base sobre la región fronto-central izquierda con presencia de puntas, ondas agudas y ráfagas de actividad rítmica de 9 Hz en dicha región con mayor negatividad en F3 (figura 3).
DISCUSIÓN
Las IRM cerebral de los casos reportados, muestran quistes porencefálicos extensos que comprometen las regiones fronto-parieto-temporales de un hemisferio diferente en cada caso. Estos hallazgos se correlacionan con la literatura existente que señala que los infartos cerebrales perinatales que resultan en quistes porencefálicos se presentan con mayor frecuencia sobre la corteza y sustancia blanca del territorio de distribución de la arteria cerebral media (ACM) (1,2).
A pesar de las impresionantes alteraciones anatómicas cerebrales presentes en los portadores de porencefalia, las manifestaciones clínicas pueden ser leves y manifestarse sólo en la etapa tardía de la infancia, siendo los más frecuentes el déficit motor leve o moderado, déficit cognitivo, crisis epilépticas, alteraciones del lenguaje, trastornos del comportamiento, defecto visual homónimo y cefaleas con características de migraña (6,7). De los casos reportados, ambos presentaban hemiparesia espástica congénita, retardo mental y epilepsia y solo la paciente mujer manifestó tener cefaleas episódicas con características de migraña.
Las crisis epilépticas de inicio focal con alteración de conciencia de ambos casos tienen características electro-clínicas y datos de neuroimágenes y evaluación neuropsicológica que permiten localizar la zona epileptogénica en la corteza dorsolateral del lóbulo frontal yacente al quiste porencefálico. La semiología de las crisis epilépticas asociadas a los quistes porencefálicos pueden ser de inicio focal con o sin alteración de la conciencia y evolución a crisis tónico- clónico bilateral (7). Naef describió diversos perfiles electro-clínicos de las crisis, incluyendo crisis focales motoras, versivas, psicomotoras y generalizadas (6).
Los registros de EEG revelaron un enlentecimiento continuo de la actividad hemisférica y descargas epileptiformes focales en región frontal del hemisferio cerebral donde se encontraba el quiste porencefálico. En otros reportes, sólo la mitad de pacientes tenían actividad epileptiforme focal, la mayoría presentaban actividad epileptiforme multifocal sobre la localización de los quistes y en otros se observaron descargas epileptiformes difusas o incluso contralaterales (8). Las anormalidades epileptiformes generalizadas del EEG son comunes en la epilepsia relacionada con la porencefalia(9). Las descargas de complejos punta- onda lenta bilateralmente sincrónicas, cuando no están acompañadas de descargas epilépticas independientes del hemisferio “sano”, pueden representar sincronía bilateral secundaria procedente del hemisferio porencefálico (7).
Según la literatura, la zona epileptogénica puede encontrarse sobre la lesión quística o localizarse en un sitio distante sobre el mismo hemisferio, o en el hemisferio contralateral (8,9). Por ejemplo, en series de casos de epilepsia asociada a porencefalia y más aún en aquellos que evaluaron epilepsia refractaria, se demostró que la esclerosis temporal mesial a menudo coexistía con la porencefalia y era el foco probable de las crisis según datos electro-clínicos concordantes (10,11).
Los pacientes que reportamos tenían crisis mensuales cuando no tomaban los fármacos antiepilépticos regularmente y quedaron libre de crisis, en forma sostenida por más de un año, cuando ellos y sus familiares fueron conscientes de las bondades del tratamiento regular. Esta respuesta al tratamiento médico es los más frecuente en epilepsia asociada a porencefalia ya que solo un 6-7% desarrollan epilepsia fármaco-resistente (7,9,12). Otro aspecto a resaltar al momento de evaluar el resultado terapéutico en ambos casos es la ausencia de patología dual entre porencefalia y esclerosis del hipocampo.
La aparición de hemiparesia congénita en dos generaciones de esta familia sugiere una predisposición genética hereditaria, sin embargo, al momento no contamos con estudios genéticos que nos permitan determinar la presencia de mutaciones genéticas asociadas a porencefalia familiar hereditaria que confirmen el diagnóstico.
En conclusión, estamos frente a dos hermanos con porencefalia familiar probable, quienes presentan hemiparesia congénita, retardo mental y epilepsia medicamente tratable con hallazgos electro- clínicos, de neuroimágenes y neuropsicológicos que permitieron localizar la zona epileptogénica sobre la región dorsolateral del lóbulo frontal yacente al quiste porencefálico. Consideramos que en pacientes con hemiparesia congénita, retardo mental y epilepsia con quiste porencefálico se debe considerar la posibilidad de porencefalia familiar e indagar antecedentes familiares de este trastorno.
REFERENCIAS BIBLIOGRÁFICAS
1. Osborn AG, Preece MT. Intracranial cysts: radiologic- pathologic correlation and imaging approach. Radiology. 2006;239: 650-64. [ Links ]
2. Govaert P. Prenatal stroke. Semin Fetal Neonatal Med. 2009;14: 250-66. [ Links ]
3. Berg RA, Aleck KA, Kaplan AM. Familial porencephaly. Arch Neurol. 1983;40:567-9. [ Links ]
4. Mancini GM, de Coo IF, Lequin MH, Arts WF. Hereditary porencephaly: clinical and MRI findings in two Dutch families. Eur J Paediatr Neurol. 2004; 8:45-54. [ Links ]
5. Meuwissen ME, Halley DJ, Smit LS, Lequin MH, Cobben JM, de Coo R, et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med. 2015;17:843-53. [ Links ]
6. Naef RW. Clinical features of porencephaly: a review of thirty-two cases. Arch Neurol Psychiatry. 1958;80:133-47. [ Links ]
7. Guzzetta F, Battaglia D, Di Rocco C, Caldarelli M. Symptomatic epilepsy in children with poroencephalic cysts secondary to perinatal middle cerebral artery occlusion. Childs Nerv Syst. 2006; 22: 922-30. [ Links ]
8. Iida K, Otsubo H, Arita K, Andermann F, Olivier A. Cortical resection with electrocorticography for intractable porencephaly-related partial epilepsy. Epilepsia. 2005; 46:76-83. [ Links ]
9. Shimizu M, Maeda T, Izumi T. The differences in epileptic characteristics in patients with porencephaly and schizencephaly. Brain Dev. 2012;34:546-52. [ Links ]
10. Ho SS, Kuzniecky RI, Gilliam F, Faught E, Bebin M, Morawetz R. Congenital porencephaly and hippocampal sclerosis: Clinical features and epileptic spectrum. Neurology. 1997; 49: 1382-8. [ Links ]
11. Burneo JG, Faught E, Knowlton RC, Martin RC, Bebin M, Morawetz R, et al. Temporal lobectomy in congenital porencephaly associated with hippocampal sclerosis. Arch Neurol. 2003; 60: 830-4. [ Links ]
12. Carreño M, Kotagal P, Perez Jiménez A, Mesa T, Bingaman W, Wyllie E. Intractable epilepsy in vascular congenital hemiparesis: clinical features and surgical options. Neurology. 2002; 59: 129-31. [ Links ]
Correspondencia:
Walter F. De la Cruz
Av. Javier Prado 1010, Torre B, Piso 3, San Isidro, Lima
Correo electrónico: walter.de.la.cruz.r@upch.pe
Teléfono: 511-294-9812
Declaración de financiamiento y de conflictos de intereses
Fuente de financiamiento: Autofinanciado. Conflictos de interés: Los autores declaran no tener ningún conflicto de interés.
Recibido: 09/05/2017
Aceptado: 15/10/2017

