










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista de Neuro-Psiquiatría
versión impresa ISSN 0034-8597
Rev Neuropsiquiatr vol.81 no.2 Lima abr. 2018
http://dx.doi.org/https://doi.org/10.20453/rnp.v81i2.3337
ARTÍCULO DE REVISIÓN
Síndrome de Lennox Gastaut. Aproximación diagnóstica y avances terapéuticos: Fármacos antiepilépticos, Canabidiol y otras alternativas.
Lennox Gastaut Syndrome. Diagnosis approach and therapeutics advances: Antiepileptic drugs, cannabidiol and other alternatives.
Manuel L. Herrera 1,a, Jorge G. Burneo 2,b
1 Hospital Edgardo Rebagliati Martins, Departamento de Neurología. Lima, Perú.
2 Epilepsy Program, Western University. London, Ontario, Canada.
a Médico Residente de Neurología; b Médico Neurólogo-Epileptólogo, Co-Director.
RESUMEN
El Síndrome de Lennox Gastaut es una encefalopatía epiléptica catastrófica de inicio en la infancia con características electro-clínicas definidas de la siguiente manera: 1) presencia de múltiples tipos de crisis epilépticas especialmente tónicas; 2) deterioro cognitivo asociado a cambios conductuales; 3) presencia de complejos punta- onda lenta generalizados en el electroencefalograma (EEG) y paroxismos generalizados de actividad rítmica rápida durante el sueño. Su etiología puede ser estructural o genética (antes denominadas sintomáticas y criptogénicas, respectivamente). El diagnóstico inicial puede ser difícil ya que con frecuencia no se identifican todos los criterios al comienzo del cuadro y el diferencial considera otros síndromes epilépticos de inicio en la infancia, tales como las epilepsias mioclónicas. El tratamiento es muy complejo, se carece de guías definidas de práctica clínica, por lo cual la experiencia de expertos es relevante. Se sugiere inicio de medicación con valproato. Lamotrigina, felbamato, topiramato, rufinamida y clobazam son los fármacos de elección de segunda línea aprobados por la Administración Federal de Alimentos y Drogas de los Estados Unidos. (FDA). El manejo quirúrgico incluye cirugía resectiva y callosotomía total o parcial. Otras alternativas son estimulación del Nervio Vago, dieta cetogénica, estimulación cerebral profunda y el uso médico de cannabis.
PALABRAS CLAVE: Lennox-Gastaut, epilepsia refractaria, crisis tónicas, paroxismos de actividad rápida, canabidiol.
SUMMARY
Lennox Gastaut syndrome is a catastrophic childhood-onset epileptic encephalopathy that presents a variety of electroclinical features: 1) Multiple seizure types, particularly tonic ones; 2) Cognitive impairment associated with behavioral disturbances; 3) Slow spike-wave complex on electroencephalographic (EEG) recordings, and generalized fast rhythms during sleep. The syndrome’s etiology can be structural or genetic. Diagnosis at the time of clinical onset may be a challenge as not all criteria are met and there may not be a full picture; the differential diagnosis should consider childhood-onset myoclonic epilepsies. Treatment is equally complex as there are no clinically practical guidelines, reason for which experts’ opinions must be sought. Initial treatment with valproic acid is suggested. Lamotrigine, felbamate, topiramate and clobazam are second line agents approved by the USA Food and Drug Administration (FDA). Surgical management may include resective surgery and/or corpus callosotomy (complete or partial). Other alternatives include Vagus Nerve Stimulation (VNS), ketogenic diet, Deep Brain Stimulation and Cannabis-based treatment.
KEYWORDS: Lennox-Gastaut, refractory epilepsy, tonic seizure, fast rhythms, Cannabidiol.
INTRODUCCIÓN
El síndrome de Lennox Gastaut (SLG) es una encefalopatía epiléptica de inicio en la infancia, definida por las siguientes características electro-clínicas: múltiples tipos de crisis, entre las que necesariamente debe haber crisis tónicas; detenimiento y/o regresión cognitiva (encefalopatía epiléptica moderada a severa y persistente en la adultez) asociado o no a desórdenes conductuales; y presencia en el trazado de EEG de los típicos complejos punta-onda lenta interictales (slow spike-wave complex), además -según algunos autores- de la presencia abrupta de ritmos de alta frecuencia (>10 Hz) durante el sueño no REM como criterio esencial (1,2).
El inicio de las crisis en SLG suele ser entre 1-8 años con un pico entre los 3-5 años. Afecta a varones y mujeres en una proporción de 5 a 1. SLG es identificado en el 4% de todas las epilepsias de inicio en la infancia (3,4). El 30% de pacientes con Síndrome de West pueden evolucionar a SLG, y de manera inversa se ha visto que un 15 a 30% de pacientes con SLG tuvieron como antecedente síndrome de West (3). La prevalencia en Europa y Norteamérica oscila entre 2-10 por 100 000 habitantes (3). No existe información sobre la epidemiologia de este síndrome en Latinoamérica.
En un 75% de pacientes con SLG se puede identificar una causa estructural subyacente (sintomáticos), estas incluyen injurias cerebrales estáticas como la encefalopatía hipóxico-isquémica, complejo de esclerosis tuberosa o síndromes neurocutáneos (TSC1 [Tuberous sclerosis complex 1] y TSC2), meningoencefalitis, malformaciones del desarrollo cortical (LIS1 [Liscencefalia tipo 1], DCX [Double cortex relation genes], GPR56 [G protein- coupled receptor]), tumores (como el hamartoma hipotalámico), hipertensión intracraneal idiopática; o injurias progresivas como los errores innatos del metabolismo (déficit de biotinidasa, desórdenes del metabolismo de creatina, entre otros) que a pesar de ser poco frecuentes tienen gran importancia por ser susceptibles de recibir tratamiento (2,4-7).
En 25 a 30% de casos la causa no es determinada (criptogénica) y se presume que son mutaciones de novo, las responsables. Reportes de casos proponen un rol etiológico en mutaciones de genes como ABRB3, ALG13, SCN8A, STXBP1, DNM1, FOXG1 o CHD2 (2,4-7).
El propósito del presente artículo fue revisar los conceptos del estado del arte respecto al diagnóstico clínico y electroencefalográfico, además de las recomendaciones actualizadas en la terapia farmacológica como en la no farmacológica.
Manifestaciones clínicas
La asociación de múltiples tipos de crisis epilépticas, con un patrón específico de EEG y deterioro intelectual son criterios diagnósticos de LGS.
Tipos de crisis
Crisis tónicas
Es el tipo de crisis más característico del SLG, al punto, que es necesaria su presencia para establecer este diagnóstico. Sin embargo, puede no estar presente al inicio del cuadro, lo que puede retrasar o diferenciar el diagnóstico. Reportes de casos como el de Chevrie y col, demuestran variedad en el momento de presentación de estas crisis, lo que está en función de la edad de los pacientes tomados como muestra (8). Por otro lado, se tiene una alta incidencia de estas crisis en aquellos estudios en que sistemáticamente se obtuvieron registros de electroencefalografía (EEG) durante el sueño (8,9).
Una crisis tónica hace referencia a un incremento sostenido de la contractura muscular que dura generalmente segundos. Puede haber variaciones de crisis tónicas como las del subtipo axial, caracterizadas por una flexión de la cabeza y tronco asociado a periodo de apnea y en ocasiones precedida de un breve llanto, quejido o grito. Cuando a este tipo de variante además se asocia la elevación y abducción de las extremidades superiores (en semiflexión o extensión), con hiperflexión de las muñecas y contractura de los puños, y piernas en extensión, se denomina crisis axo- rizomélica (9,10). Cuando hay compromiso de casi la totalidad de la musculatura incluidas las porciones distales, se denominan crisis tónicas globales. Ocasionalmente, puede haber crisis tónicas muy sutiles en las que las únicas manifestaciones clínicas pueden ser gesticulaciones y posturas tónicas transitorias del tronco y de las extremidades proximales; inclusive puede haber sólo una supraversión ocular lenta aislada (9,10).
Cuando el paciente está de pie, la flexión de extremidades inferiores o la flexión del tronco pueden precipitar caídas que pueden hacer difícil la tipificación de las crisis, pudiendo confundirla con una crisis atónica, inclusive mioclónica (1).
Ausencias atípicas
Es el segundo tipo de crisis más frecuente en SLG, se caracterizan por una pérdida progresiva y en ocasiones parcial de la conciencia, de breve duración (segundos) y de recuperación progresiva. Estas crisis suelen darse en pacientes con un grado de retardo intelectual significativo por lo que puede resultar difícil su identificación clínica debido al grado de reactividad que se encuentra previamente disminuido. Estas crisis pueden acompañarse en ocasiones de babeos, cambios en el tono postural y mioclonías palpebrales y periorales, lo que puede confundirlas con crisis focales con alteración de conciencia o con crisis generalizadas con patrón de punta onda en el EEG (8,9).
Drop attacks (Crisis de caídas)
Son caídas bruscas debidas a crisis tónicas súbitas, crisis atónicas, crisis mioclónicas o crisis mioclónica- atónica. Ocurren en el 56% de pacientes con SLG (8) y se caracterizan por pérdidas muy breves del tono postural (0,5 a 0,8 s) que generan caídas hacia adelante o hacia atrás de la cabeza o de todo el cuerpo y son responsables de lesiones significativas y recurrentes que pueden generar o acentuar la incapacidad del paciente. Son las crisis tónicas súbitas las principales responsables de estas caídas por la falta de equilibrio resultante de la contracción simultánea de músculos agonistas y antagonistas de un segmento o de toda la superficie corporal (1).
Crisis mioclónicas
Son crisis muy breves (<100 ms) pero que pueden precipitar caídas. Están asociadas por lo general a síndromes epilépticos generalizados, por tanto, su hallazgo debe tomarse como una característica asociada más no diagnóstica en SLG. Existe una “variante mioclónica” que se da en el 18% de pacientes con SLG que se caracteriza por ser de inicio tardío, tener predominio de crisis mioclónicas respecto a las tónicas (incluso pueden no tenerlas o ser muy sutiles durante el sueño), menor deterioro intelectual y tener mejor pronóstico (1)
Estado epiléptico no convulsivo
Aproximadamente el 50 a 75% de pacientes con SLG presentan estos episodios que pueden durar de horas a días y que se caracterizan por un estado subcontinuo de crisis de ausencia atípica con un grado variable de compromiso del nivel de conciencia y que es interrumpido periódicamente por breves crisis tónicas recurrentes. La frecuencia y duración de estos episodios de estado tienen relación proporcional directa con el deterioro cognitivo (1).
Otras crisis
Además de las crisis más frecuentes mencionadas, se pueden dar otros tipos: crisis focales con o sin compromiso bilateral posterior, crisis tónico-clónicas generalizadas, crisis clónicas, etc. Por lo general estas crisis adicionales se dan en estados más avanzados de la enfermedad, aunque ocasionalmente pueden aparecer en su inicio dificultando más el diagnóstico.
Deterioro Intelectual, discapacidad o deterioro cognitivo
Es el otro componente de la triada diagnóstica clásica. En aquellos pacientes con SLG sintomático se encuentra un grado variable de deterioro cognitivo y/o del desarrollo al momento del inicio del síndrome clínico (60-70% de los casos) y que suele ser de moderado a severo y que se autolimita en un determinado momento (1-4,12).
En los pacientes con SLG de etiología criptogénica (20-30%) se halla por lo general un desarrollo normal hasta el momento en que las crisis aparecen (inicio tardío) y generan un cuadro de regresión intelectual de grado variable (2,4).
Hay un incremento del deterioro cognitivo del 75 al 95% por cada 5 años desde el inicio del síndrome (11). Hay un 10-20% de niños que cursan con un nivel cognitivo dentro del rango de normalidad pero que suelen tener dificultades en las actividades de la vida diaria que parecen ser causadas por un enlentecimiento en el procesamiento mental (1,11).
Evaluación diagnóstica
Debido a lo amplio de su definición, el diagnóstico de SLG puede ser dado erróneamente al denominar como tal a cuadros epilépticos con múltiples tipos de crisis refractarias y de inicio en la infancia, como en los desórdenes en que predominan las crisis mioclónicas- astáticas para los cuales las metas de tratamiento son muy distintas a los de pacientes con SLG.
La incertidumbre de la nosología, la ausencia de las crisis típicas al inicio del cuadro, y el hecho de que los patrones de EEG como los complejos punta-onda lenta no son patognomónicos del SLG dificultan aún más su diagnóstico inicial.
El diagnóstico de SLG depende de la presencia (o su desarrollo en el tiempo) de los criterios electro- clínicos antes definidos. La presencia de crisis tónicas y los patrones de paroxismos de actividad rápida son probablemente los hallazgos más característicos de este síndrome.
Hallazgos en electroencefalograma
Características del EEG interictal
El clásico hallazgo en el EEG son los paroxismos de complejos punta-onda lentos (POL) (10). Estos se pueden hallar aislados o en número variable, constan de una punta (<70 ms) o una onda aguda (70 – 200 ms) seguidos de una depresión profunda positiva y luego de una onda negativa (300 – 400 ms) (figura 1).
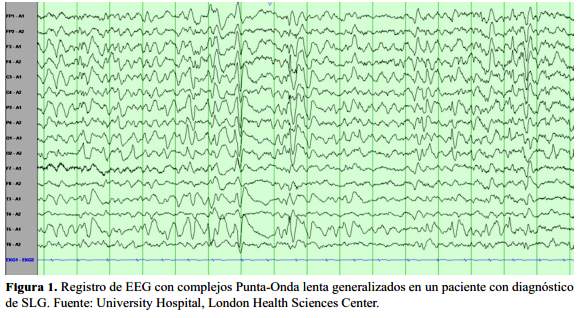
Estos complejos son bilaterales y sincrónicos con una frecuencia de 1-2 Hz. Tienen mayor amplitud en las regiones frontales y fronto-centrales. A diferencia de las POL de las crisis de ausencia, las que se aprecian en el SLG varían en frecuencia, amplitud y morfología de un paroxismo a otro, o incluso en el mismo paroxismo. En descargas paroxísticas prolongadas, los POL tiene una frecuencia mayor a los 3 Hz vistos en registros durante la vigilia o el sueño no REM (1,9,12).
Aunque los complejos POL pueden asociarse a descargas ictales, se hallan en muchos casos en el registro interictal y por tanto no tienen una traducción clínica. La gran cantidad de POL, su aumento o decremento, dificultan la diferenciación entre los patrones ictales e interictales. Los complejos POL de más de 3 Hz dan un correlato clínico cuando duran más de 3s. Adicionalmente la estimulación luminosa no desencadena los complejos POL, hecho que distingue al SLG de las epilepsias mioclónicas. Sin embargo, la apertura ocular, el llamado del paciente por su nombre, el estímulo doloroso, disminuyen los paroxismos de POL; mientras que la relajación y la somnolencia lo incrementan. (1,12,13).
Durante el sueño no REM aumenta significativamente la aparición de POL hasta en el 95% de los casos (13-15); por el contrario, durante la fase de sueño REM, disminuyen. Es prudente recalcar que la fase de sueño REM es muy escasa o nula en los pacientes con SLG (13).
Características del EEG Ictal
Patrones súbitos de ritmos rápidos (>10 Hz) bilaterales o difusos o polipuntas, paroxismos generalizados de actividad rápida rítmica, son registradas durante el sueño no REM. Estas descargas súbitas duran pocos segundos (1-9 s), pero tienden a recurrir a intervalos breves, y por lo general, son idénticas, pero más cortas que las descargas vistas en crisis tónicas, que tienen un ritmo de reclutamiento -de baja amplitud al inicio con un aumento gradual de la amplitud-. Estas descargas pueden parecer no tener un correlato clínico evidente en ocasiones, pero la exploración simultánea con poligrafía y electromiografía revela periodos breves de apnea y contracción leve de musculatura axial, respectivamente. Por lo tanto, es necesario el registro de EEG durante el sueño no REM para identificar su presencia. Esta actividad rápida paroxismal generalizada se encuentra en un 80% de pacientes con SLG a diferencia del 15% que se encuentra en pacientes con crisis focales con sincronía bilateral secundaria, por lo que resulta útil en la discriminación entre ambos (1,13-14) (figura 2).
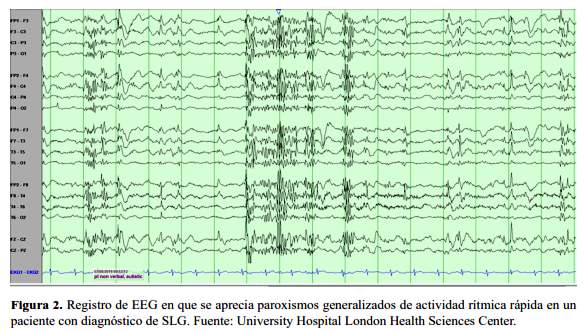
EEG en crisis tónicas
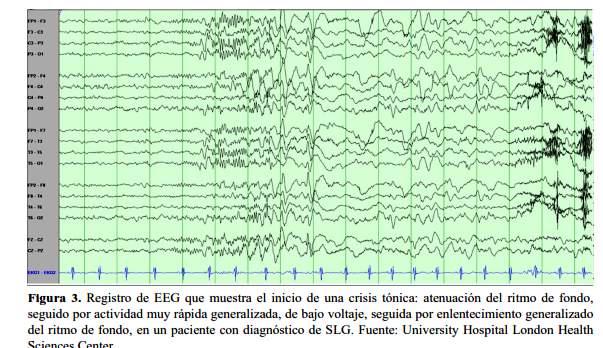
El ritmo de reclutamiento es lo característico, con una actividad rápida generalizada de baja amplitud inicialmente y con aumento gradual posterior, pasando de 50 uV hasta 100 Uv; y de una disminución en su frecuencia; aunque en ocasiones puede haber descargas rítmicas de 10-15 Hz de alta amplitud desde el inicio. Puede estar precedido de puntas, ondas agudas o hipovoltaje con aplanamiento del registro. Posteriormente hay un periodo corto de actividad delta antes de retornar al patrón interictal de POL (1,13-14) (figura 3).
EEG Ausencias Atípicas
Se observan complejos POL similares a los del trazado interictal, pero de mayor amplitud, además de ser más regulares y sostenidos, bisincrónicos y simétricos, por lo general de menos de 2,5 Hz; a diferencia de las puntas, ondas agudas o incluso polipuntas que se pueden hallar en la descarga ictal de las ausencias típicas, que tienen una frecuencia mayor a 3 Hz en la infancia, y de hasta 6 Hz en la adolescencia (1, 13).
EEG en Drop Attacks
El patrón ictal EEG es heterogéneo y está en función del tipo de crisis subyacente. En las crisis tónicas súbitas breves puede no haber cambios o un ligero aplanamiento en el trazado; mientras que en los de mayor duración (> 1s) puede haber paroxismos de actividad rápida de baja amplitud. En las producidas por crisis mioclónicas hay paroxismos de POL, polipuntas, o polipunta-onda lenta generalizados que se correlacionan con la sacudida mioclónica (8,16).
EEG en Estado Epiléptico no convulsivo
El EEG puede ser muy similar al trazado interictal con persistencia de POL en el trazado o verse anormal al punto de exhibir un patrón ipsarrítmico atípico (13).
Estudios complementarios
El estudio con neuroimágenes suele ser muy útil para determinar alteraciones estructurales cerebrales como malformaciones corticales (lisencefalia, polimicrogiria, neoplasias, esclerosis tuberosa, encefalopatía hipóxica-isquémica, etc.) (2,4).
Los estudios de neuroimagen funcional como la RMN funcional, la Tomografía por emisión de positrones (PET), la tomografía computarizada de emisión monofotónica (SPECT), adquieren relevancia en el estudio prequirúrgico para la mejor delimitación del foco epileptogénico a reseccionar (3).
El estudio genético debe incluir la hibridación genómica comparada para identificar variaciones en la estructura y número de copias con respecto a todo el genoma humano; o realizar una secuenciación específica de genes en función a la sospecha diagnóstica (5-7).
DIAGNÓSTICO DIFERENCIAL
El diagnóstico de SLG depende de la correlación electroclínica, como se mencionó antes, con los criterios ya desarrollados. Las características más representativas del SLG son la predominancia de crisis tónicas y el patrón de ritmo rápido durante el sueño. Sin embargo, al inicio del cuadro puede no encontrarse estos hallazgos y el debut puede ser con otro tipo de crisis, o el patrón interictal POL de EEG durante la vigilia puede estar ausente, razón por la que toma gran relevancia el registro de EGG durante el sueño para identificar los patrones paroxísticos de actividad rítmica rápida (1,8,13).
En la práctica clínica es frecuente que los síndromes epilépticos no tengan una diferencia clara en su inicio y en algunos casos precedan al desarrollo de otro, como en el caso de un síndrome de West, con espasmos e hipsarritmia que progresa a un cuadro de SLG con POL y diversos tipos de crisis (20% de los casos de SLG) (3, 14-16). Diferenciar espasmos prolongados de crisis tónicas puede ser complejo y se debe tener presente que los primeros tienden a repetirse en “salvas o racimos”, a diferencia de las segundas en donde la contracción muscular es sostenida por tiempo variable (11-13).
Entre las epilepsias de inicio en la infancia, el síndrome de Dravet (Epilepsia Mioclónica severa de la Infancia) es un cuadro que cursa con deterioro cognitivo, con descargas generalizadas de puntas y ondas, hallazgos similares a los del SLG. Sin embargo, en este cuadro es frecuente el antecedente de crisis febriles prolongadas focales (hemiclónicas) o generalizadas durante el primer año de vida. Después, entre 1-4 años aparecen crisis mioclónicas o de ausencia atípica, principalmente (las crisis tónicas o los espasmos son muy infrecuentes, al punto que de existir debe dudarse de este diagnóstico). A diferencia del SLG, el EEG anormal de Dravet se exacerba con el sueño, con la deprivación y con maniobras de activación como la fotoestimulación y no hay presencia de paroxismos generalizados de actividad rítmica rápida (9-13) (tabla 1).
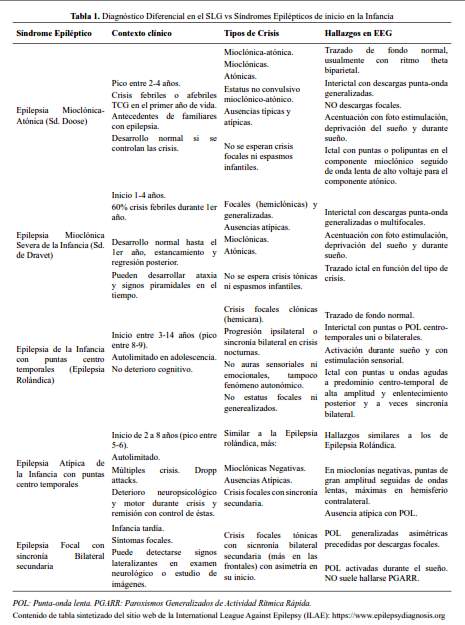
La presencia entre los 2 y 5 años de crisis mioclónicas y atónicas, en un niño que por lo general no tiene deterioro intelectual, un registro de EEG con puntas-ondas generalizadas rápidas o lentas con actividad theta biparietales debe hacer sospechar de un Síndrome de Doose (Epilepsia Mioclónica-Astática o Epilepsia con Crisis Mioclónicas-Atónicas), al que en una etapa posterior pueden añadirse crisis tónicas y deterioro cognitivo si no se controlan las crisis (11-13) (tabla 1).
Un diagnóstico diferencial importante es con la Epilepsia Atípica de la Infancia con puntas centrotemporales, llamada también Síndrome de Pseudo Lennox, o Epilepsia Parcial Benigna Atípica (antiguamente). Es un cuadro que se desarrolla en niños de 2-8 años con un pico entre 4-6 y que se caracteriza por ser autolimitado y asentar en niños sin previo deterioro cognitivo, pero con antecedentes en su mayoría de crisis febriles o afebriles (11-13). Las principales crisis son las focales motoras clónicas (fronto-parieto operculares) con usuales movimientos clónicos de hemicara y que durante los episodios nocturnos pueden extenderse a la extremidad superior ipsilateral, a la contralateral y llegar a tener sincronía bilateral generalizada. Las otras crisis características son las mioclónicas negativas que pueden conllevar a episodios de múltiples caídas (gran actividad de crisis por semanas) seguidos de periodos libres de crisis de incluso meses. Puede haber crisis de ausencias atípicas durante la vigilia. El EEG registra puntas y ondas lentas generalizadas durante el sueño a predominio centro-temporal. No hay paroxismos de actividad rápida rítmica durante el sueño y no se espera apreciar crisis tónicas a diferencia del SLG (11-13) (tabla 1).
TRATAMIENTO
Manejo farmacológico
Resultados de ensayos
Se han realizado ensayos clínicos aleatorizados controlados, en pacientes con el cuadro ya establecido de encefalopatía epiléptica, para determinar la eficacia y seguridad de algunos fármacos antiepilépticos (FAE) como lamotrigina, felbamato, topiramato, clobazam, rufinamida; fármacos que han sido aprobados por la FDA para su uso en SLG (17-24). Postura sustentada en una revisión de Cochrane de estudios aleatorizados controlados que concluyó que lamotrigina y felbamato redujeron significativamente la frecuencia de todos los tipos de crisis respecto a placebo (32% vs 9% [P=0,02]; y 19% vs 4% [P=0,002], respectivamente) (17,18). Respecto a la reducción de Drop Attacks, se halló que lamotrigina, topiramato y felbamato tuvieron significancia estadística (17-20).
Felbamato tiene muchos mecanismos de acción incluido su antagonismo por los receptores de N-metil- D-aspartato. En un estudio clase I de 73 pacientes con SLG, felbamato redujo significativamente todos los tipos de crisis (19% vs 4% del placebo) (18,19). Su uso se ve limitado por sus peligrosos efectos colaterales como anemia aplásica (1 en 5 000) y falla hepática (1 en 26 000), ésta última aumenta con el uso concomitante de valproato a 1 en 500 en niños menores de 2 años y 1 en 12 000 en adultos (19).
Respecto al control de crisis tónicas, se encontró que lamotrigina y felbamato redujeron de manera significativa la frecuencia de crisis (17,18). No se halló repercusión significativa para el caso del topiramato (20). El impacto sobre el control de las crisis de ausencia atípica no pudo ser estimado por el diseño de la revisión, y las características de las mismas crisis (difícil identificación y alta frecuencia) (17-20).
Con respecto a la rufinamida, droga que había caído en el desuso, se halló que tenía una reducción muy significativa en la frecuencia total de crisis versus placebo (32,7% a 11,7% con P= 0,0015); y en la reducción de las crisis tónico-atónicas versus placebo (42,5% vs 1,4% con un P<0,0001) (24).
En relación al ácido valproico, estudios no controlados describen que se obtiene un control de más del 50% de las crisis en: 55% de pacientes con drop attacks; 25-30% de pacientes con crisis de ausencia atípica y Mioclónicas; y menor tasa de respuesta para las crisis tónicas y tónico-clónicas (25,26). Sus principales efectos adversos son la hepatotoxicidad y la trombocitopenia que puede empeorar con la politerapia.
En otros estudios pequeños no controlados, se halló que las benzodiacepinas tenían una mejora en el control de crisis en el 46-70% de pacientes (26).
Son los fármacos más usados en forma aguda, y debe evitarse su uso crónico, por desarrollar tolerancia, somnolencia, incremento de secreciones bronquiales y orales, riesgo de crisis por supresión o riesgo de crisis tónicas, incluso estatus, por uso frecuente. Dentro de este grupo, el clobazam es el fármaco de elección para terapia de adición, por tener un perfil de mayor seguridad: menor somnolencia, menor interacción medicamentosa, rápido inicio de acción, vida media más larga, amplio espectro; sin embargo, puede incrementar la espasticidad en pacientes con daño neurológico extenso perinatal (21-23).
El clobazam es uno de los pocos fármacos cuyo uso en Perú fue reportado en al menos una serie de casos (20 pacientes con epilepsias refractarias, de los cuales 11 tenían el diagnóstico de Lennox Gastaut) por Patricia Campos, del Hospital Cayetano Heredia en el año 1993; en cuya casuística (con seguimiento en un plazo no menor a tres años) se evidenciaba la seguridad y el mejor perfil de este fármaco en el manejo coadyuvante de estos síndromes epilépticos y se proponía por tanto su uso (27). (Se sugería al final del estudio hacer una referencia a la realidad nacional, que escapa un tanto al tema de revisión global del SLG, y que se dificulta por la falta de guías de práctica clínica en centros avanzados por lo que se apela a la recomendación de expertos, eso sumado a la falta de reporte de experiencia nacional en el manejo de estos pacientes).
La carbamazepina y la oxcarbazepina pueden ser utilizadas para el control de crisis tónico clónica generalizadas (CTCG) y crisis focales (principalmente focales estructurales), pero tiene especial utilidad en el control de las crisis tónicas, pero su empleo requiere una vigilancia estricta para detectar posibles efectos adversos o el agravamiento de otros tipos de crisis como las ausencias atípicas, las mioclónicas y el estatus epiléptico no convulsivo (26,28).
Levetiracetam ha demostrado adecuado perfil de seguridad y eficacia en series de casos como la reportada por De los Reyes y Sharp. Esta droga se ha convertido en un fármaco de común elección por el perfil descrito y la tolerabilidad (28).
La etosuximida tiene gran relevancia para el control de las crisis de ausencia atípica y mejora también el control de las crisis atónicas y mioclónicas negativas (26). El fenobarbital y primidona empeoran los problemas conductuales, aumentan el deterioro cognitivo y son más sedantes (26,29). El fenobarbital
pueda exacerbar las crisis de ausencia (29). La fenitoína es útil para el control de CTCG, pero agrava las mioclónicas y ausencias atípicas (26).
Respecto al uso de esteroides y la hormona adrenocorticotropa (ACTH), hay reportes aislados que sugieren beneficio en el control de las ausencias atípicas, drop attacks y estatus epiléptico no convulsivo; sin embargo, no hay evidencia suficiente que sustente su uso a largo plazo (además de los efectos adversos y la supresión del eje hipotálamo- hipófisis-adrenal) y se reserva como terapia de rescate en periodos de exacerbación del cuadro (29,30). El uso de Inmunoglobulina humana aún está en estudio; no obstante, hay algunos resultados preliminares alentadores como el de Ferrie et al., (29,30).
Dentro de los efectos adversos de los fármacos antiepilépticos (FAE), tanto en su uso aislado como del producto de su interacción, el potencial para exacerbar una crisis es una consideración que no se debe soslayar; por ejemplo, el uso de benzodiacepinas puede predisponer al desarrollo de crisis tónicas cuando se administran en el intento de controlar otros tipos de crisis (26,33). Por lo mencionado es importante determinar correctamente el tipo de crisis y/o síndrome epiléptico para escoger el FAE adecuado en la dosis correcta y tener en cuenta las interacciones con drogas administradas concomitantemente (33).
Consideraciones generales del manejo
El manejo de un paciente con SLG resulta altamente complejo por muchas razones como la refractariedad de las crisis durante la evolución del cuadro; la falta de animales de experimentación en que se pueda reproducir el fenómeno patológico subyacente para su posterior estudio; la gran variedad de crisis que se presenta y que obliga a elegir FAE que pueden controlar un tipo pero exacerbar otro; además de la politerapia que se instala con el aumento de riego de reacciones adversas y de interacciones medicamentosas (26).
Las múltiples variables que convergen en un paciente con SLG dificultan seriamente la realización de ensayos clínicos que generen evidencia de clase I o II para las terapias múltiples. Hancock et al., condujeron una revisión en Cochrane con la finalidad de hacer un meta-análisis y concluyeron que no era posible realizarlo por la heterogeneidad de las poblaciones y múltiples variables estudiadas (26,34). Al momento se dispone de la experiencia clínica de expertos como Monica Lemmon y Eric Kossoff, del departamento de Neurología del Hospital Johns Hopkins, que orientan el abordaje terapéutico de estos casos (34).
Una encuesta realizada a 57 expertos en epilepsia pediátrica en Europa sobre la aproximación al manejo de pacientes con SLG evidenció el uso de Ácido Valproico como fármaco de primera línea (36). Si no hubiese control adecuado con este primer fármaco, el
57% de ellos recomienda añadir un segundo fármaco para lograr el control de crisis, mientras que un 38% recomienda cambiar el fármaco (lamotrigina) y persistir en monoterapia (36).
Similar recomendación se obtuvo en una encuesta parecida realizada en USA, en donde la primera opción seguía siendo el ácido Valproico o la lamotrigina (38). El topiramato se sugirió como segunda opción (37).
Es vital informar al familiar sobre las limitaciones del tratamiento, los aumentos o disminuciones que puede haber en la frecuencia de las crisis, el riesgo de reacciones adversas y la interacción medicamentosa que aumenta con la polifarmacia (1,26).
Es importante tratar las comorbilidades asociadas como los problemas conductuales y psiquiátricos (déficit de atención con hiperactividad, ansiedad, depresión conducta agresiva, psicosis, etc.,) que requieren una atención integral (1).
La respuesta clínica o la presencia de reacciones adversas será el parámetro para mantener, modificar o cambiar de FAE. Diversos estudios sustentan este juicio, al encontrarse respuestas óptimas con el mismo fármaco, pero con niveles séricos muy variables (26).
Manejo no farmacológico
Tratamiento quirúrgico
El manejo quirúrgico en pacientes con epilepsias refractarias se ha convertido en la aproximación más exitosa para el control de crisis a largo plazo (26).
La cirugía resectiva beneficia puede beneficiar a aquellos pacientes con lesiones focales como las displasias, hamartomas o infartos. En un estudio de cohorte retrospectivo de 27 niños y adolescentes con SLG (85% con epilepsia lesional por MRI) se realizó cirugía resectiva y se obtuvo un 60% de pacientes libres de crisis, y una mejora significativa en otro 15% en un seguimiento a 33 meses (38).
La callosotomía parcial o total es el tratamiento quirúrgico paliativo primario en los pacientes con SLG que tienen alta frecuencia de crisis que conllevan a drop attacks. Se ha documentado disminución significativa en la frecuencia de las crisis tónicas y atónicas (hasta un 80%) (39). En las crisis tónico- clónicas la tasa de respuesta es menor. Se debe replantear el riesgo/beneficio en aquellos pacientes con un moderado desarrollo cognitivo y analizar las variables de su adaptación social, conducta motora y funcionamiento intelectual (26,38,39).
La estimulación del Nervio Vago (VNS, de sus siglas en inglés) es un procedimiento que ha demostrado utilidad en la reducción de los drop attacks. Tiene menos riesgo de comorbilidad que la callosotomía y se puede utilizar como terapia coadyuvante a ésta. Frost M y colaboradores, en diferentes estudios prospectivos abiertos, encontraron que tras 6 meses de estimulación en pacientes con SLG hubo una reducción del total de crisis de 46-58%, y específicamente las crisis atónicas se redujeron en un 88% (40). Adicionalmente en el seguimiento a largo plazo de los pacientes con VNS no desarrollaron efectos adversos, es más, se reportó en algunos casos cambios conductuales positivos (40). Un meta-análisis que comparó callosotomía corporal (CC) vs VNS halló que en el primer grupo hubo una reducción de >50% de las crisis atónicas en el 80% de pacientes vs un 54% de pacientes con VNS con un P<0,05. Todas las otras crisis fueron estadísticamente similares (41).
Dieta cetogénica
La dieta cetogénica ha demostrado eficacia en múltiples síndromes epilépticos (3). Aunque su mecanismo de acción no es totalmente comprendido, se sabe que disminuye la excitabilidad neuronal a través de la reducción de carbohidratos, activación de los canales de potasio, inhibición de la vía de la rapamicina e inhibición neuronal mediada por el glutamato. Se describe mejor control de la frecuencia total de crisis en algunos pacientes, específicamente en las crisis atónicas y mioclónicas, en las que se obtiene una reducción de más del 50% de estas (42).
Estimulación cerebral profunda
Estudios preliminares sobre estimulación eléctrica cerebral profunda (DBS de sus siglas en inglés) a nivel de los núcleos talámicos centromediales, reportan disminución significativa en la frecuencia de crisis en pacientes con epilepsia generalizada refractaria a fármacos y cirugía. En un ensayo clínico controlado simple ciego, realizado en dos centros: King’s College Hospital (London, U.K.) y el Hospital Universitario La Princesa (Madrid, Spain) se encontró que la DBS fue útil en los pacientes con epilepsia generalizada mas no en los pacientes con epilepsia focal frontal y que en el primer grupo, conformado por 6 pacientes, hubo una disminución de más del 50% de las crisis en 5 de ellos (un paciente libre de crisis, otro con reducción del 99% de crisis y en los otros tres hubo reducción entre el 65 y 90% de crisis); por lo que concluyen en que DBS parece ser un procedimiento seguro y eficaz en pacientes con epilepsia generalizada refractaria (43). Velasco Al y col. describen una reducción general del 80% de las crisis en 13 pacientes con SLG asociado a una mejora en el grado de independencia de los cuidadores (43).
Uso del Cannabis sativa (Marihuana)
Existía el antecedente del uso del Cannabis medicinal desde tiempos muy remotos para diversos desórdenes de salud, entre ellos la epilepsia; pero fue durante las primeras décadas del siglo XX, con el descubrimiento del fenobarbital que se dejó de lado su uso masivo. Sin embargo, actualmente ha recobrado importancia como posibilidad terapéutica en los casos de epilepsias refractarias debido a diversos factores como: el mayor conocimiento del sistema de canabinoides endógenos del Sistema Nervioso Central (SNC) que sustenta la idea de un potencial efecto anticonvulsivo de los canabinoides sintéticos (44); la presión social impulsada por la necesidad de los familiares de los pacientes aquejados con esta condición de hallar una alternativa de manejo frente al panorama poco alentador; reportes de casos aislados, series de casos, ensayos clínicos iniciales y factores ajenos al ámbito científico (44,45).
Existen más de 500 sustancias activas en las especies de Cannabis, de las cuales las más abundantes son los canabinoides, y de esta familia, dos son los compuestos más estudiados por ser los más neuroactivos: el Tetrahidrocanabinol (THC) y el Canabidiol (CBD); el primero con propiedades más psicoactivas y asociado a deterioro cognitivo en su uso crónico, por lo que los esfuerzos actuales están dirigidos en el estudio con ensayos clínicos controlados randomizados con compuestos de alta pureza para CBD como el Epidiolex (44).
Un reciente ensayo clínico abierto dirigido por Devinsky et al., realizado en pacientes con epilepsia refractaria de inicio en la infancia para valorar la seguridad y eficacia del CBD de alta pureza (Epidiolex) como terapia aditiva a regímenes antiepilépticos previos, tuvo como resultado que hubo una media de
37% en la reducción de crisis motoras y que hubo una reducción >50% del total de las crisis en el 37% de pacientes (45). Hubo también una reducción media del
69% de los drop attacks en los pacientes identificados con crisis atónicas. Estos hallazgos sugirieron que el CBD es seguro y eficaz en el manejo de esta patología y que se requieren ensayos clínicos controlados y aleatorizados para determinar el verdadero perfil de seguridad y eficacia (45).
Datos recientes de un ensayo clínico doble ciego aleatorizado dirigido por Thiele et al., hallaron que el uso de CBD como terapia aditiva a regímenes previos de FAE reducía el total de crisis en una media del 45% respecto al 15% del placebo (46). Los drop attacks se redujeron en un 49% comparado con el 20% del grupo con placebo (46). Serios efectos adversos ocurrieron en 23% de pacientes que recibieron CBD vs el 5% con placebo (46). Sus resultados sugieren que es seguro y eficaz el uso de CBD como terapia aditiva para el control de drop attacks en pacientes con epilepsia refractaria por SLG (46).
PRONÓSTICO
La evolución de los pacientes con síndrome de Lennox Gastaut suele ser desfavorable y existen factores de mal pronóstico identificados como la etiología estructural (sintomática), que haya sido precedido por el Síndrome de West, que haya un inicio temprano (< 3 años), que haya alta frecuencia de crisis y estatus epilépticos y la persistencia de la actividad lenta de base o de los complejos POL generalizados (3,4).
El pronóstico sigue siendo malo para los pacientes con SLG por la refractariedad a la que suelen evolucionar sus crisis, los déficits cognitivos y conductuales que inevitablemente se desarrollan en grado variable por el síndrome clínico en sí mismo y por efectos adversos sumados de la politerapia antiepiléptica, por las comorbilidades derivadas de esta condición, el aumento de hospitalizaciones por estatus epilépticos y el mayor riesgo de muerte súbita inexplicable relacionada a la epilepsia (SUDEP) (3,15,16).
En conclusión, el Síndrome de Lennox Gastaut es una encefalopatía epiléptica catastrófica, con múltiples tipos de crisis, asociado a deterioro cognitivo conductual y patrón electroencefalográfico característico en la mayoría de casos, que sigue siendo un reto respecto a su fisiopatología, al diagnóstico temprano y a su terapéutica.
REFERENCIAS BIBLIOGRÁFICAS
1. Arzimanoglou A, French J, Blume W, Cross H, Ernst JP, Feucht M, et al. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol.2009; 8: 82-93. [ Links ]
2. Mastrangelo M. Lennox–Gastaut Syndrome: A state of the art review. Neuropediatrics. 2017; 48(3):143-151. [ Links ]
3. Resnick T, Sheth R. Early diagnosis and treatment of Lennox-Gastaut Syndrome. J Child Neurol. 2017;32(11):947-955. [ Links ]
4. Muradi H, Doaa K, Mohammed M. Lennox-Gastaut síndrome. Neurosciences. 2015; 20(3): 207–212. [ Links ]
5. Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty N, et al. De novo mutations in epileptic encephalopathies. Nature. 2013; 501 (7466): 217-21. [ Links ]
6. Terrone G, Bienvenu T, Germanaud D, Barthez- Carpentier M, Bertrand D, Delanoe C, et al. A case of Lennox- Gastaut syndrome in a patient with FOXG1-related disorder. Epilepsia. 2014; 55(11): 116–119. [ Links ]
7. Lund C, Brodtkorb E, Oye AM, Rosby O, Selmer KK. CHD2 mutations in Lennox-Gastaut syndrome. Epilepsy Behav. 2014; 33:18–21. [ Links ]
8. Chevrie J, Aicardi J. Childhood epileptic encephalopathy with slow spike-wave. A statistical study of 80 cases. Epilepsia. 1972; 13: 259-71. [ Links ]
9. Gastaut H, Broughton R. Tonic seizures. In: Gastaut H, Broughton R. Epileptic seizures. Springfield, Illinois: CC Thomas; 1972.p. 37–47. [ Links ]
10. Blume W, Luders H, Mizrahi E, Tassinari C, Emde Boas W, Engel J. ILAE Commission Report. Glossary of descriptive terminology for ictal semiology: report of the ILAE task force on classification and terminology. Epilepsia. 2001; 42: 1212–18. [ Links ]
11. Aicardi J, Chevrie JJ. Atypical benign epilepsy of childhood. Dev Med Child Neurol. 1982; 24 (3): 281-92. [ Links ]
12. Bourgeois B, Douglass L, Sankar R. Lennox-Gastaut syndrome: a consensus approach to differential diagnosis. Epilepsia. 2014; 55 (4): 4–9. [ Links ]
13. Dulac O, N’Guyen T. The Lennox-Gastaut syndrome. Epilepsy 1993; 34 (Suppl. 7): S7-S17. [ Links ]
14. Gastaut H, Zifkin BG. Secondary bilateral synchrony and Lennox Gastaut syndrome. In: Niedermeyer E, Degen R, (Eds). The Lennox Gastaut syndrome. New York: Alan R Liss; 1988.p. 221–42. [ Links ]
15. Beaumanoir A. The Lennox-Gastaut syndrome. In: Roger J, Bureau M, Dravet C, (Eds.) Epileptic syndromes in infancy, childhood, and adolescence. London: John Libbey; 1985.p.89-99. [ Links ]
16. Markand O. Lennox-Gastaut syndrome (childhood epileptic encephalopathy). J Clin Neurophysiol. 2003; 20: 426-441. [ Links ]
17. Motte J, Trevathan E, Arvidsson J, Barrera M, Mullens E, Manasco P. Lamotrigine for generalized seizures associated with Lennox-Gastaut Syndrome. N Engl J Med. 1997; 337: 1807-12. [ Links ]
18. The Felbamate Study Group in Lennox-Gastaut Syndrome. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). N Engl J Med. 1993; 328: 29-33. [ Links ]
19. Pellock JM, Faught E, Leppik IE, Shinnar S, Zupanc ML. Felbamate: consensus of current clinical experience. Epilepsy Res. 2006;71(2-3):89–101. [ Links ]
20. Sachdeo R, Glauser T, Ritter F, Reife R, Lim P, Pledger G. A double-blind, randomized trial of topiramate in Lennox-Gastaut syndrome. Neurology. 1999; 52:1882-87. [ Links ]
21. Munn R, Farrell K. Open study of clobazam inrefractory epilepsy. Pediatr Neurol. 1993; 9: 465-69. [ Links ]
22. Jan M, Shaabat A. Clobazam for the treatment of intractable childhood epilepsy. Saudi Med J. 2000; 21: 622–24. [ Links ]
23. Vassella F, Pavlincova E, Schneider H, Rudin H, Karbowski K. Treatment of infantile spasms and Lennox-Gastaut syndrome with clonazepam (Rivotril). Epilepsia. 1973; 14: 165–75. [ Links ]
24. Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox-Gastaut syndrome. Neurology. 2008; 71: 1950-58. [ Links ]
25. Covanis A, Gupta A, Jeavons P. Sodium valproate: monotherapy and polytherapy. Epilepsia. 1982; 23: 693-720. [ Links ]
26. Ostendorf A, Yu-Tze N. Treatment-resistant Lennox- Gastaut syndrome: therapeutic trends, challenges and future directions. Neuropsychiatric Disease and Treatment. 2017;13: 1131–1140. [ Links ]
27. Campos P. Uso de clobazam en epilepsias de difícil control en niños. Arq Neuro-Psiquiatr. 1993; 51 (1): 66-71. [ Links ]
28. De Los Reyes E, Sharp G, Williams J, Hale S. Levetiracetam in the treatment of Lennox-Gastaut syndrome. Pediatr Neurol. 2004; 30: 254–56. [ Links ]
29. Panayiotopoulos C. A clinical guide to epileptic syndromes and their treatment: Based on the ILAE classifications and practice parameter guidelines. 2nd ed. London: Springer; 2010. [ Links ]
30. Ferrie C, Patel A. Treatment of Lennox-Gastaut Syndrome (LGS). Eur J Paediatr Neurol. 2009;13(6):493–504. [ Links ]
31. VanStraten A, Ng Y. Update on the management of Lennox-Gastaut syndrome. Pediatr Neurol.2012;47(3):153–161. [ Links ]
32. Zaccara G. The treatment of epilepsy. In: Shovon SD, Perucca E, Engel J Jr, editors. The Treatment of Epilepsy. Oxford: John Wiley & Sons; 2009.p.459-474. [ Links ]
33. Schmidt D, Bourgeois B. A risk-benefit assessment oftherapies for Lennox-Gastaut syndrome. Drug Saf. 2000; 22: 467–77. [ Links ]
34. Lemmon M, Kossoff E. New treatment options for Lennox-Gastaut syndrome. Current Treat Options Neurol. 2013;15(4):519-528. [ Links ]
35. Van Rijckevorsel K. Treatment of Lennox-Gastaut syndrome: overview and recent findings. Neuropsych Dis Treat. 2008; 4: 1001-1019. [ Links ]
36. Wheless J, Clarke D, Arzimanoglou A, Carpenter D. Treatment of pediatric epilepsy: European expert opinion, 2007. Epileptic Disord. 2007; 9: 353–412. [ Links ]
37. Wheless JW, Clarke DF, Carpenter D. Treatment of pediatric epilepsy: expert opinion, 2005. J Child Neurol. 2005; 20: S1–56. [ Links ]
38. Lee Y, Kang H, Lee J, Kim S, Kim D, Shim K, et al.Resective pediatric epilepsy surgery in Lennox-Gastaut syndrome. Pediatrics. 2010;125(1): e58–e66. [ Links ]
39. Gates J. Surgery in Lennox-Gastaut syndrome: Corpus collosum division for children. Adv Exp Ed Biol. 2002; 497: 87–98. [ Links ]
40. Frost M, Gates J, Helmers S, Wheless J, Levisohn P, Tardo C, et al. Vagus nerve stimulation in children with refractory seizures associated with Lennox- Gastaut syndrome. Epilepsia. 2001; 42: 1148–52. [ Links ]
41. Lancman G, Virk M, Shao H, Mazumdar M, Greenfield J, Weinstein S, et al. Vagus nerve stimulation vs corpus callosotomy in the treatment of Lennox-Gastaut syndrome: a meta-analysis. Seizure. 2013;22(1):3–8. [ Links ]
42. Freeman J, Vining E. Seizures decrease rapidly after fasting: preliminary studies with the ketogenic diet. Arch Pediatr Adoles Med. 1999; 53: 946–49. [ Links ]
43. Velasco A, Valesco F, Jimenez F, Velasco M, Castro G, Carrillo-Ruiz J, et al. Neuromodulation of the centromedian thalamic nuclei in the treatment of generalized seizures and the improvement of the quality of life in patients with Lennox-Gastaut syndrome. Epilepsia. 2006; 47: 1203–12. [ Links ]
44. Friedman D, Devinsky O. Cannabinoids in the treatment of epilepsy. N Engl J Med. 2015; 373:1048-58. [ Links ]
45. Devinsky O, Eric M, Daniel F, Thiele E, Laux L, Sullivan J, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol. 2016; 15: 270–78. [ Links ]
46. Thiele E, Marsh E, French J, Mazurkiewicz- Beldzinska M, Benbadis S, Joshi C, et al. Cannabidiol in patients with seizures associated with Lennox- Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018; 391(10125):1085-1096. [ Links ]
Declaración de financiamiento y de conflicto de intereses: El presente artículo de revisión no requirió financiamiento de institución alguna.
Los autores declaran no tener conflictos de intereses.
Correspondencia:
Manuel Herrera Aramburu
Hospital Nacional Edgardo Rebagliati Martins. Departamento de Neurología Piso 13A. Av Edgardo Rebagliati 490
Jesús María, Lima, Perú.
Teléfono: 511265 4901 / 51 992194265
Correo electrónico: herrera4a@hotmail.com
Recibido: 01/11/2017
Aceptado: 08/05/2018

