










Services on Demand
Journal

Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkRevista de Neuro-Psiquiatría
Print version ISSN 0034-8597
Rev Neuropsiquiatr vol.81 no.4 Lima Oct./Dec. 2018
http://dx.doi.org/10.20453/rnp.v81i4.3439
ARTÍCULO DE REVISIÓN
Epilepsia mioclónica juvenil
Juvenile myoclonic epilepsy
David J. Caldera 1,a, Jorge G. Burneo 2,b.
1 Universidad Católica Redemptoris Mater. Managua, Nicaragua.
2 Programa de Epilepsia, Western University, London Health Sciences Center. Ontario, Canadá.
a Medico General; b Profesor de Neurología.
RESUMEN
La epilepsia mioclónica juvenil (EMJ) es un trastorno generalizado que se inicia usualmente en la pubertad o adolescencia y se caracteriza por la presencia de mioclonías y, con menor frecuencia, crisis tónico-clónica generalizadas y ausencias. A nivel internacional, se estima que anualmente tiene lugar un nuevo caso de EMJ por cada 1000-2000 personas. El diagnóstico es fundamentalmente de naturaleza clínica, corroborado por información electroencefalográfica. El fármaco de primera elección para el tratamiento de la EMJ sigue siendo el ácido valpróico; sin embargo, se han reportado resultados eficaces con lamotrigina y levetiracetam para el control de EMJ en monoterapia o politerapia, con topiramato como terapia coadyuvante para el control de las crisis tónico-clónicas generalizadas.
PALABRAS CLAVE: Epilepsia, epilepsia mioclónica juvenil, mioclonía.
SUMMARY
Juvenile Myoclonic Epilepsy (JME) is a generalized type of epilepsy characterized by the occurrence of myoclonic seizures and, less frequently, of generalized tonic-clonic seizures and absences. The onset usually occurs during puberty or adolescence. Worldwide, it is estimated that there is a new case of JME per year for every 1000-2000 people. Its diagnosis is fundamentally clinical, corroborated by EEG tests. The first drug of choice for the treatment of JME is still valproic acid; however, lamotrigine and levetiracetam have shown efficacious results for the control of JME, used as monotherapy or polytherapy with topiramate as a coadyuvant for the control of generalized tonic- clinic seizure.
KEY WORDS: Epilepsy, juvenile, myoclonic epilepsy, myoclonus, JME
INTRODUCCIÓN
La epilepsia mioclónica juvenil (EMJ), tambien denominado por Janz y Christinas como el sindrome de Petit mal impusivo de Janz, es clasificada como una epilépsia generalizada genetica, con fenotipos variables (1), a este tipo de epilepsia se le define la presencia de sacudidas mioclónicas, sin alteración del conocimiento y con ocurrencia generalmente después del despertar (2,3).
Las crisis tónico-clónica generalizadas pueden también ocurrir, y en menor frecuencia las ausencias. Estas se ven a menudo precipitadas por la falta de sueño, al momento de despertar (4).
El objetivo del presente trabajo es el de revisar la epilepsia mioclónica juvenil, incluida su epidemiología, presentación clínica y pronostico, avances en la neuroimagen, genética y su tratamiento.
Epidemiologia
La incidencia de EMJ en la población general se estima en 1 caso por cada 1000-2000 personas por año (5). La EMJ representa aproximadamente el 5-10% de todas las epilepsias; sin embargo, las cifras exactas pueden ser más altas, ya que a menudo la enfermedad se diagnostica erróneamente (5).
Los pacientes con EMJ no suelen presentar déficits intelectuales o neurológicos y la enfermedad sigue un curso no progresivo (6).
Presentación clínica
La identificación de crisis mioclónicas generalizadas es fundamental para el diagnóstico de EMJ (7). La edad de inicio de la EMJ es alrededor de los 10 y 25 años (8). Las mioclonías se caracterizan por sacudidas únicas o repetitivas, abruptas, arrítmicas e involuntarias, que afectan predominantemente los hombros y los brazos; generalmente simétricos, sin embargo, pueden ser unilaterales, la mioclonía más típica es la elevación del hombro con flexión del codo. Las crisis mioclónicas generalizadas son breves, se describe una duración de hasta un segundo con una fase de relajación más lenta; la amplitud varía de movimientos violentos a contracciones mínimas (9). En algunos casos, no hay movimientos visibles, y el paciente informa solo una sensación subjetiva de choque eléctrico dentro del cuerpo. A este fenómeno se le conoce como mini-polimioclonías (7).
Se describe la participación de las extremidades superiores en el 97,7% de los pacientes, mientras que en menor frecuencia en las extremidades inferiores (46,5%), tronco (23,3%) y cara (14%) (10). La mayoría de las mioclonías ocurren al despertar (62,8-87,5%) (8,9). El 3-5% de los pacientes con EMJ solo tienen crisis mioclónicas (7). Las crisis tónico-clónica generalizadas (CTCG) están presentes en el 80-97% de los pacientes (11), y aparecen meses o años después de la aparición de mioclonías (6). Ocurren después de despertar en el 53,7% de los casos (10). A menudo son precedidos por crisis mioclónicas generalizadas leves a moderadas, de frecuencia e intensidad crecientes (7). Las ausencias están presentes entre el 12 al 54% (11).
Los factores desencadenantes de los ataques epilépticos son variados, siendo el más frecuente la privación del sueño, reportada hasta en un 58.3-89.5% de los casos (11, 12). Otros factores desencadenantes incluyen la fatiga (73,7%), estímulos visuales luminosos (36.8%), menstruación (24,1%) concentración mental (22,8%) y estrés, excitación o frustración (12.3%). También se encontró que la ingestión de alcohol es un factor desencadenante en el 51,3% de los casos (10).
Estudios paraclínicos Electroencefalograma (EEG)
El diagnóstico de la EMJ es electro-clínico, basado en la presencia de las características clínicas ya mencionadas. El electroencefalograma (EEG) provee un apoyo; en el EEG, la EMJ típicamente presenta un ritmo de fondo dentro de los límites de la normalidad (6). El hallazgo interictal incluye descarga de puntas y complejos punta-ondas generalizados y poli puntas- y-ondas, la frecuencia puede ser hasta de 6Hz y de dominio frontocentral (figura 1 y figura 2) (13).
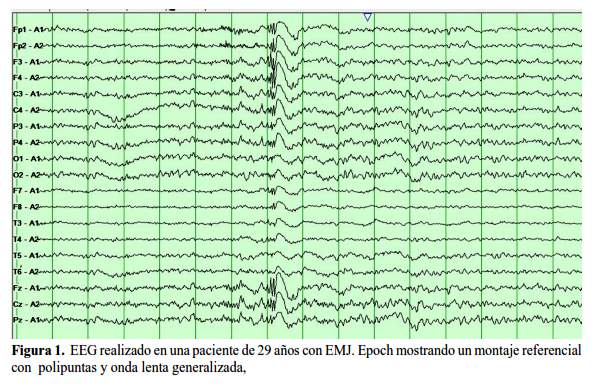
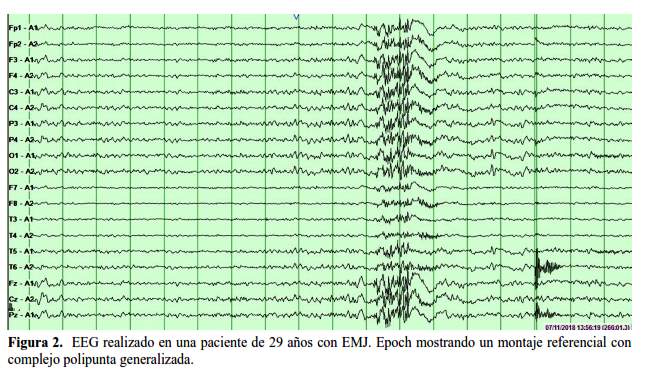
El típico EEG ictal de una crisis mioclónica muestra el patrón de poli puntas-y-ondas, que consiste en un grupo de 5 a 20 puntas generalizadas, casi siempre simétricas y de alta frecuencia, generalmente seguidas por ondas lentas en una frecuencia de 2.5-5 Hz (10,11,14). Se pueden observar otros patrones en los EEG ictales, como la punta-onda generalizada. También se pueden encontrar ondas agudas y lentas, ráfagas de ondas lentas y/o ondas agudas (14).
La crisis mioclónica ocurre simultáneamente con el patrón poli punta-onda, pero este último continúa más allá de la terminación de la mioclonía y puede persistir durante varios segundos (9).
Estimulación fótica intermitente
Durante esta maniobra se puede objetivar la presencia de foto sensibilidad, la cual se define como respuesta anormal a la luz solar o artificial producida por reactividad extrema de las moléculas que absorben la luz en los tejidos. La respuesta fotoparoxística que se puede encontrar en pacientes con EMJ, se caracteriza por el registro con descargas epileptiformes tipo puntas o complejos punta-onda usualmente generalizadas, que ocurren durante la maniobra de foto estimulación. Es el correlato electroencefalográfico de la fotosensibilidad (15,16). Se desencadena con mayor frecuencia con flashes a 10-20 Hz (16,17). Presenta máxima penetrancia entre los 5-20 años, con mayor énfasis en púberes y adolescentes entre 11-15 años, decrece después de los 20 años y ocurre principalmente en mujeres (15-19).
Estudios de neuroimagen
Las técnicas de neuroimagen recientemente desarrolladas han detectado anomalías estructurales. Aunque en la EMJ no se han encontrado anomalías en la resonancia de encéfalo estructural convencional de 1.5 T (11,20).
Resonancia Magnética
La resonancia magnética (RM)estructural del cerebro no suele evidenciar anomalías. Esta observación refleja el hecho de que la EMJ es una epilepsia generalizada genética y no es causada por afecciones que conducen a una patología cerebral cortical focal, como tumores cerebrales o encefalitis. Sin embargo, los estudios morfométricos cuantitativos que utilizan una técnica basada en voxel han mostrado algunas diferencias entre los pacientes con EMJ (5). Estos hallazgos no tienen una significancia clínica clara.
A pesar de estas pequeñas diferencias cuantitativas, las guías emitidas por la Liga Internacional contra la Epilepsia (ILAE) no recomiendan estudios de neuroimagen de rutina en pacientes con EMJ (21).
Genética
Se han descubierto 15 loci ligados a la EMJ, siendo el más importante el dominio Myoclonina1/ EF (terminal C)-EFHC1, el cual está presente en 3-9% de familias con ésta epilepsia. Mutaciones de EFHC1 pueden causar pequeñas malformaciones del desarrollo de la corteza (22).
Por otro lado, el estudio histopatológico de Meencke y Janz continúa siendo controversial. Los autores encontraron micro-disgenesis en 3 pacientes con EMJ de un grupo de 15 pacientes con epilepsia genética generalizada (23). Lamentablemente, estos hallazgos no han sido replicados.
Finalmente, existe evidencia de estudios en familias con EMJ (24), que existen anormalidades del comportamiento, que al parecer también estuvieron genéticamente determinadas, sugiriendo que la EMJ es un desorden más complejo del desarrollo neuronal (25).
Diagnóstico diferencial
Una razón para el diagnóstico inadecuado es que los pacientes a menudo no informan las crisis mioclónicas o el médico no pregunta de manera adecuada al obtener la información de las crisis. En la mayoría de los pacientes, la EMJ se diagnostica después de que el paciente experimenta una crisis tónico-clónico generalizado (CTCG) y ha comenzado a tratarse con uno de los fármacos habituales de primera línea, como la fenitoína o la carbamazepina (5). El reconocimiento que la Carbamazepina exacerba el cuadro clínico debe conducir a un diagnóstico y terapia apropiados. Las convulsiones mioclónicas no generan daño, pero pueden generar daño físico si el paciente se cae durante la crisis.
Las condiciones a considerar en el diagnóstico diferencial incluyen lo siguiente: Síndrome de epilepsia generalizada con CTCG solamente, epilepsias mioclónicas progresivas, epilepsia de ausencia juvenil y epilepsia del lóbulo frontal.
Tratamiento
El tratamiento se basa en el equilibrio entre evitar los factores desencadenantes y el uso adecuado de fármacos antiepilépticos (FAE).
El estilo de vida tiene una parte muy importante en el tratamiento. Este incluye recomendaciones como evitar desencadenantes comunes, incluyendo la supresión del sueño, fatiga, ingesta de alcohol, despertarse de manera inoportuna y énfasis en la importancia de la adherencia al tratamiento con FAE.
Desde 1980, el ácido valproico (AVP) se ha considerado el FAE de primera elección en la EMJ, con una tasa de respuesta de hasta el 80% (26). Su utilidad no solo está basada en su efecto en las crisis mioclónicas, sino también en las CTCG y las ausencias que también pueden estar presentes. Los pacientes con EMJ a menudo responden a dosis bajas de AVP consistente en 1000 mg diarios o menos (27). En la actualidad, el AVP es la primera elección de FAE en hombres con EMJ. Pero, la evidencia sugiere evitar el uso de AVP en mujeres en edad fértil, debido a que significativamente, dependiendo de la dosis, aumenta el riesgo de malformaciones fetales y deterioro del desarrollo cognitivo postnatal, incluido el trastorno del espectro autista (28, 29).
Durante los últimos años, se han sugerido lamotrigina y topiramato como opciones alternativas (30). La monoterapia con lamotrigina resultó en una tasa libre de ataques del 40% al 83% para pacientes con EMJ (31). La monoterapia con lamotrigina y terapia combinada con topiramato fueron opciones efectivas para el tratamiento de pacientes en los cuales no se puede usar el AVP. Datos de varias series pequeñas y grandes describen que la lamotrigina se puede usar como mono- o politerapia en pacientes con EMJ (32-
37), mientras que el topiramato generalmente como politerapia. El tratamiento se dirigió principalmente a las CTCG en pacientes con epilepsia genética generalizada, incluidos algunos pacientes con EMJ (31). Las CTCG y las crisis de ausencia se controlan en casi el 80% de los pacientes, y las crisis mioclónicas se controlan en un 72% (31).
El topiramato fue altamente efectivo para las CTCG, aunque no tanto para las crisis mioclónicas (36). El número de pacientes con EMJ tratados con topiramato en monoterapia en varias series es demasiado bajo como para hacer un comentario definitivo sobre si la monoterapia con topiramato puede ser una opción efectiva. En un informe de consenso, el AVP, lamotrigina y topiramato se clasificaron como el primer, segundo y cuarto fármaco de elección, respectivamente. El clonazepam, que ocupó el tercer lugar, es útil para las crisis mioclónicas y tiene un papel menor en el tratamiento de las crisis de GTC (38).
El estudio realizado por Prasad et al., demostró que: el ácido valproico, la lamotrigina y el tratamiento con topiramato individualmente no demostraron ninguna diferencia significativa en los resultados de CTCG, mioclonías y crisis de ausencia (31). Los 3
FAE produjeron un buen control de las crisis de GTC (68% - 80%), las crisis de ausencia (50% - 78%) y las crisis mioclónicas (58% - 59%). Sin embargo, los datos combinados del control de ataques mioclónicos por todos los FAE no fueron óptimos cuando se compararon con los datos del control de CTCG.
En algunos casos, la lamotrigina no ofrece un adecuado control de las crisis mioclónicas. Se cree que esto es debido a su acción bloqueante del canal de sodio, similar al mecanismo de acción de la fenitoína y la carbamazepina, que pueden empeorar el control de las ausencias y las crisis mioclónicas en pacientes con epilepsia genética generalizada.
El levetiracetam (LEV), uno de los recientes FAE, presenta amplias ventajas por su absorción sin cambios con los alimentos y en la porfiria aguda, especialmente en pacientes en edad fértil, en pacientes con epilepsia y espectro autista, en pacientes con mioclonías corticales de diverso origen (39). En países de América del Norte y Oceanía se utiliza más LEV qué AVP en la EMJ (40). La tolerabilidad de esta droga fue buena en todos los estudios. La tasa de interrupción debido a eventos adversos es baja (0-12%) (41). Los efectos secundarios observados ocurren en la primera 5 meses de tratamiento en el 17,2% al 51,3% de los pacientes, estos están relacionados con el sistema nervioso central (42) y se caracterizaron por nerviosismo e irritabilidad (40).
Las crisis convulsivas en aproximadamente el 15% de los pacientes con EMJ no responden al tratamiento con FAE. Este grupo, al parecer se caracteriza por tener una historia de muchas crisis convulsivas antes del inicio del tratamiento médico, o que tienen una respuesta inadecuada al tratamiento inicial (43). Si bien este grupo de pacientes no son candidatos a cirugía resectiva, debido a su naturaleza generalizada, otros tratamientos no farmacológicos pueden ser considerados. Además, está indicado admitir a los pacientes a una unidad de monitoreo de video-EEG prolongado, para descartar una posible epilepsia focal (de origen frontal), que haya podido ser confundida con EMJ.
La neuro-estimulación es un tratamiento que se debe considerar cuando los FAEs no permiten controlar las crisis. La neuro-estimulación más usada incluye la estimulación del nervio vago (ENV). En un estudio de 12 pacientes con epilepsia generalizada, de los cuales 7 tuvieron diagnóstico de EJM, se encontró una reducción de sus crisis del 50% o más. Las CTCG se redujeron hasta en un 61%, mientras que las mioclonías en un 40% (44). El tratamiento con el ENV es relativamente seguro y altamente tolerado. En nuestra experiencia, la ronquera intermitente y la baja de peso son los efectos colaterales más comunes.
Otro tipo de tratamiento incluye las dietas especiales, entre ellas, la más estudiada ha sido la dieta cetogénica, ésta se usa en diversos tipos de epilepsia en niños, quizás con mayor frecuencia en aquellos con epilepsias difíciles de tratar, como el síndrome de Lennox-Gastaut. En adultos, la dieta cetogénica no es bien tolerada, por lo que en la mayoría de los programas de epilepsia, la dieta Atkins modificada es la más usada. La Dieta Atkins modificada es una opción menos restrictiva y más factible que la dieta cetogénica. Quizás, sea la más atractiva para aquellos con capacidad intelectual normal en general (45).
Pronóstico
En general, la EMJ ha sido considerada un tipo de epilepsia ‘benigna’ con muy buena respuesta al tratamiento con FAEs. Lamentablemente, farmacoresistencia se ha visto en cerca de 15% de pacientes. Aunque, se ha descrito que existe pseudo- farmacoresistencia hasta en un 10% de pacientes (46). La remisión a largo plazo de las crisis es más favorable de lo que se creía. Janz público su propia serie con información de seguimiento disponible hasta 69 años después (promedio 44.6 años), y encontró que 59.1% de pacientes permanecen libres de crisis al menos 5 años después del último contacto (47). En este grupo,71,8% seguían en tratamiento, mientras que 28.2% no tomaban mas medicación. En otras series los hallazgos son similares (26-28.6% sin medicación y libre de crisis) en seguimientos de hasta 20 años o más (48,49). Lamentablemente, 74% de pacientes con EMJ tuvieron al menos un evento social desfavorable, como no haber concluido la educación escolar, embrazo no planeado, depresión, desempleo o vivir solo (48).
CONCLUSIONES
La identificación de las mioclonías al despertar es fundamental para el diagnóstico de EMJ. El típico EEG de una crisis mioclónica muestra patrones de polipunta-onda, caracterizados por un grupo de puntas generalizados, simétricas y de alta frecuencia, seguidos por ondas lentas. Los estudios de neuroimagen no están indicados de manera rutinaria. Los fármacos por considerar incluyen el ácido valproico, el levetiracetam, la lamotrigina, el clonazepam y el topiramato. Donde en el caso de los varones la primera opción es AVP y en las mujeres LVT o LTG, debido al alta posibilidad de toxicidad fetal. El pronóstico en general es adecuado por varios años después de haberse iniciado el tratamiento.
REFERENCIAS BIBLIOGRÁFICAS
1.Janz D. Epilepsy with impulsive petit mal (juvenile myoclonic epilepsy). Acta Neurol Scand. 1985; 72(5):449-59. [ Links ]
2. Engel J. ILAE classification of epilepsy syndromes.Epilepsy Res. 2006; 70(Suppl 1): S5-10. [ Links ]
3. Scheffer IE, Berkovic S, Capovilla G, et al. EILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58(4):512-521. doi: 10.1111/epi.13709
4. Janz D. Juvenile myoclonic epilepsy. Epilepsy with impulsive petit mal. Cleve Clin J Med. 1989 ;56(Suppl1): S23-3. [ Links ]
5.Selph J, Sen S. Juvenile Myoclonic Epilepsy: Clinical Presentation. Bethesda: MedScape; 2016. [ Links ]
6.Arzimanoglou A, Guerrini R, Aicardi J. Epilepsies with predominantly myoclonic seizures. In: Arzimanoglou A, Guerrini R. Aicardi’s epilepsy in children. Philadelphia: Lippincott Williams & Wilkins; 2004. p. 58-80. [ Links ]
7. Renganathan R, Delanty N. Juvenile myoclonic epilepsy: under-appreciated and under-diagnosed. Postgrad Med J. 2003; 79(928): 78-80. [ Links ]
8. Yacubian E. Juvenile myoclonic epilepsy: Challenges on its 60th anniversary. Seizure. 2017; 44: 48-52. [ Links ]
9. Faught E. Clinical presentations and phenomenology of myoclonus. Epilepsia. 2003; 44: 7-12. [ Links ]
10. Hrachovy R, Frost J. The EEG in selected generalized seizures. J Clin Neurophysiol. 2006; 23(4): 312-32. [ Links ]
11. Mehndiratta M, Aggarwal P. Clinical expression and EEG features of patients with juvenile myoclonic epilepsy (JME) from North India. Seizure. 2002;11(7): 431-6. [ Links ]
12. Pedersen S, Petersen A. Juvenile myoclonic epilepsy: clinical and EEG features.Acta Neurol Scand. 1998; 97 (3): 160-3. [ Links ]
13. Sousa N, Sousa P, Garzon E, Sakamoto A. Juvenile myoclonic epilepsy: Analysis of factors implied in delayed diagnosis and prognosis after clinical and electroencephalographical characterization. J Epilepsy Clin Neurophysiol. 2005; 11: 7-13. [ Links ]
14. Sousa NA, Sousa-Pda S, Garzon E, Sakamoto AC, Braga NI, Yacubian EM. EEG recording after sleep deprivation in a series of patients with juvenile myoclonic epilepsy). Arq Neuropsiquiatr. 2005;63(2): 383-8. [ Links ]
15. Whitehead K, Sherratt M, Kandler R, Lawrence S, Pang C. Photic stimulation during electroencephalography: Efficacy and safety in an unselected cohort of patients referred to UK neurophysiology departments. Seizure. 2016;34:29-34. doi: 10.1016/j.seizure.2015.11.005
16. Grosso S, Pucci L, Bartalini G, et al. Photoparoxysmal responses in children with chromosomal aberrations. Epilepsy Res. 2006; 72(2): 164-70. [ Links ]
17. Binelli S, Ragona F, Canafoglia L, et al. Electroencephalographic (EEG) Photoparoxysmal Responses Under 5 Years of Age: Diagnostic Implications and Peculiarities. J Child Neurol. 2015;30(13): 1824-30. doi: 10.1177/0883073815583687
18. Strigaro G, Falletta L, Varrasi C, Rothwell JC, Cantello R. Overactive visuomotor connections underlie the photoparoxysmal response: A TMS study. Epilepsia. 2015; 56(11): 1828-35. [ Links ]
19. Guellerin J, Hamelin S, Sabourdy C, Vercueil L. Low- frequency photoparoxysmal response in adults: an early clue to diagnosis. J Clin Neurophysiol. 2012;29(2):160-4. doi: 10.1097/WNP.0b013e31824d949f
20. Lin K, Jackowski AP, Carrete H Jr, et al. Voxel-based morphometry evaluation of patients with photosensitive juvenile myoclonic epilepsy. Epilepsy Res. 2009; 86(2): 138-45. [ Links ]
21. Gaillard WD, Chiron C, Cross JH, et al. Guidelines for imaging infants and children with recent-onset epilepsy. Epilepsia. 2009; 50(9): 2147-53. [ Links ]
22. Delgado-Escueta AV, Koeleman BP, Bailey JN, Medina MT, Durón RM. The quest for juvenile myoclonic epilepsy genes. Epilepsy Behav. 2013;28 (Suppl 1):S52-7. doi: 10.1016/j.yebeh.2012.06.033
23. Meencke H, Janz D. The significance of microdysgenesia in primary generalized epilepsy: an answer to the considerations of Lyon and Gastaut. Epilepsia. 1985; 26(4): 368-71. [ Links ]
24. Opeskin K, Kalnins RM, Halliday G, Cartwright H, Berkovic SF. Idiopathic generalized epilepsy: lack of significant microdysgenesis. Neurology. 2000;55(8): 1101-6. [ Links ]
25. Schmitz B, Yacubian EM, Feucht M, Hermann B, Trimble M. Neuropsychology and behavior in juvenile myoclonic epilepsy. Epilepsy Behav. 2013;28 (Suppl 1):S72-3. doi: 10.1016/j.yebeh.2013.02.019
26. Penry J, Dean J, Riela A. Juvenile myoclonic epilepsy: long-term response to therapy. Epilepsia. 1989; 30 (Suppl 4): S19-23. [ Links ]
27. Hernández-Vanegas LE, Jara-Prado A, Ochoa A, et al. High-dose versus low-dose valproate for the treatment of juvenile myoclonic epilepsy: Going from low to high. Epilepsy Behav. 2016;61:34-40. doi: 10.1016/j.yebeh.2016.04.047
28. Tomson T. Commentary: Valproate in the treatment of epilepsy in women and girls: The need for recommendations. Epilepsia. 2015; 56(7): 1004-5. [ Links ]
29. Tomson T. Withdrawal of valproic acid treatment during pregnancy and seizure outcome: Observations from EURAP. Epilepsia. 2016; 57(8): 173-7. [ Links ]
30. Genton P. Juvenile myoclonic epilepsy and related syndromes: clinical and neurophysiological aspects. In: Malafosse A. Idiopathic generalized epilepsies: Clinical, experimental and genetic aspects. London: John Libby; 1994. p. 253-265. [ Links ]
31. Prasad A, Kuzniecky RI, Knowlton RC, et al. Evolving antiepileptic drug treatment in juvenile myoclonic epilepsy. Arch Neurol. 2003; 60(8): 1100-5. [ Links ]
32. Buchanan N. The use of lamotrigine in juvenile myoclonic epilepsy. Seizure. 1996; 5(2): 149-51. [ Links ]
33. Kustra R. Lamotrigine monotherapy in patients with Juvenile Myoclonic Epilepsy: focus on seizure and myoclonus freedom. Epilepsia. 2001; 41: 181-1. [ Links ]
34. Nadkarni M. Lamotrigine in treatment of juvenile myoclonic epilepsy. Epilepsia. 2001; 42: 211. [ Links ]
35. Stein A, Carrazana E. The use of lamotrigine in juvenile myoclonic epilepsy. Epilepsia. 2001; 42:261. [ Links ]
36. Biton V. Topiramate in juvenile myoclonic epilepsy: observations from randomized controlled trials in primary generalized tonic-clonic seizures. Epilepsia. 1999; 40: 226. [ Links ]
37. Rosenfeld W. A broad-spectrum agent in patients with juvenile myoclonic epilepsy. Epilepsia. 1999;41: S77-S81. [ Links ]
38. Obeid T, Panayiotopoulos C. Clonazepam in juvenile myoclonic epilepsy. Epilepsia. 1989; 30(5): 603-6. [ Links ]
39. Perla D, Lackington I. Levetiracetam. Rev Chil Epilep. 2005; 6(1): 1-4. [ Links ]
40. Wilmshurst J. Treatment of infants with epilepsy: Common practices around the world. Epilepsia. 2015;56(7): 1033-46. [ Links ]
41. Weijenberg A, Brouwer O, Callenbach P. Levetiracetam monotherapy in children with epilepsy: A systematic review. CNS Drugs. 2015; 29(5): 371-82. [ Links ]
42. Coppola G. Levetiracetam during 1-year follow-up in children, adolescents, and young adults with refractory epilepsy. Epilepsy Res. 2004; 59(1): 35-42. [ Links ]
43. Kwan P, Brodie M. Early identification of refractory epilepsy. N Engl J Med. 2000; 342(5): 314-9. [ Links ]
44. Kostov H, Larsson P, Roste G. Is vagus nerve stimulation a treatment option for patients with drug- resistant idiopathic generalized epilepsy? Acta Neurol Scand Suppl. 2007; 187: 55-8. [ Links ]
45. Giner P. Epilepsia y alimentacion: Dietas cetogenicas. Nutr Hosp Suppl. 2009; 2(2): 79-88. [ Links ]
46. Gelisse P, Genton P, Thomas P, Rey M, Samuelian JC, Dravet C. Clinical factors of drug resistance in juvenile myoclonic epilepsy. J Neurol Neurosurg Psychiatry. 2001; 70(2): 240-3. [ Links ]
47. Senf P, Schmitz B, Holtkamp M, Janz D. Prognosis of juvenile myoclonic epilepsy 45 years after onset: seizure outcome and predictors. Neurology. 2013;81(24): 2128-33. [ Links ]
48. Camfield C, Camfield P. Juvenile myoclonic epilepsy 25 years after seizure onset: a population-based study. Neurology. 2009; 73(13): 1041-5. [ Links ]
49. Geithner J, Schneider F, Wang Z, et al. Predictors for long-term seizure outcome in juvenile myoclonic epilepsy: 25-63 years of follow-up. Epilepsia. 2012;53(8):1379-86. doi: 10.1111/j.1528-1167.2012.03526.x [ Links ]
Recibido: 16/08/2018
Aceptado: 16/12/2018

